高性能计算服务
>
软件专区
>
最佳实践
>
使用RASPA2做气体吸附计算
RASPA是一款专为模拟纳米孔系统中的吸附和扩散现象而设计的通用经典模拟软件包。它通过模拟分子在孔隙结构中的运动,帮助科学家和工程师理解材料的微观行为,进而优化材料的性能。RASPA的模拟方法包括分子动力学、蒙特卡洛方法等,适用于多种类型的孔隙材料,如沸石、金属有机框架(MOFs)和多孔有机聚合物(POPs)。
RASPA的主要贡献者包括来自不同研究机构的科学家和工程师,David Dubbeldam, Sofia Calero, Thijs J. H. Vlugt, Donald E. Ellis, Randall Q. Snurr
RASPA具备以下功能特点:
本文,我们将介绍如何使用RASPA软件计算多孔材料的气体等温吸附曲线,阐述巨正则系综蒙特卡洛方法的基本原理,并展示如何在超算互联网使用这一软件。
等温吸附曲线描述了在一定温度下,吸附质在吸附剂上的吸附量与吸附质平衡压力(或浓度)之间关系的曲线。一些多孔的物质,例如沸石分子筛、金属有机框架材料(MOFs)、共价有机框架材料(COFs),以及一些无定形多孔碳、岩石中的干酪根有机质,均可以吸附气体分子,例如水分子、CO2分子、CH4分子、烃类分子等。
根据在指定温度下的气体吸附曲线,可以得到在不同压强状态下,多孔材料对相应气体的吸附特点、最大吸附量等性质,在实际应用中,研究人员通过气体吸附模拟,可以与实验中测量结果进行比对,分析吸附机理,辅助材料设计。
巨正则系综是统计物理中的概念,其中,系综是一个巨大的系统,由组成、性质、尺寸和形状完全一样的全同体系构成、数目极多的系统的集合,其中每个系统各处在某一微观运动状态,而且是各自独立的。组成系综的系统与一温度为T、化学势为μ的很大的热源、粒子源相接触,此时系统不仅和热源有能量交换,而且可以同粒子源有粒子的交换,最后达到平衡,这种系综称巨正则系综(μVT)。
气体吸附问题涉及气体分子在吸附剂结构中的插入、删除和移动,大量的蒙特卡洛模拟后体系达到平衡的状态,则可以得到气体吸附量。巨正则系综蒙特卡洛模拟的Metropolis方法实现如下:
(1) 根据系统设定(粒子数、体积、温度),产生初始构象;
(2)由旧(初始)构象出发,随机产生新的构象(以移动粒子为例)
(3)根据新旧状态的能量差ΔE=U(rnew)-U(rold),判断新状态的取舍,判断舍选有以下情况:
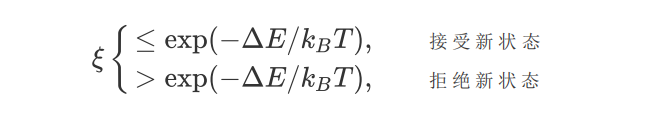
如果新状态被拒绝,则把原来的状态作为新状态,重复进行步骤2,并记录一次。
除了移动粒子操作,巨正则系综还涉及粒子的插入或删除操作:
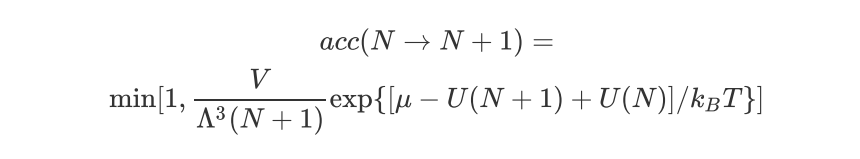
删除一个粒子的接受概率是
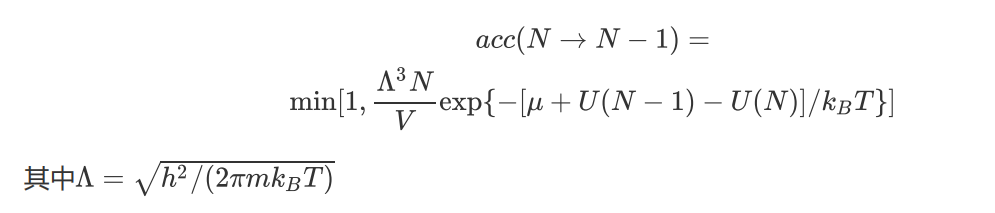
超算互联网为您提供了多种版本、多种编译方式、链接库全面的RASPA软件包,满足您的需求。这里我们详细介绍基于超算互联网 Raspa2 v2.0.48版本进行GCMC计算。
登录超算互联网https://www.scnet.cn, 搜索"RASPA",找到您所需的版本,选择计算资源(若无计算资源,需要先购买资源)、同意服务协议、去使用软件,等待软件部署完毕即可使用软件。
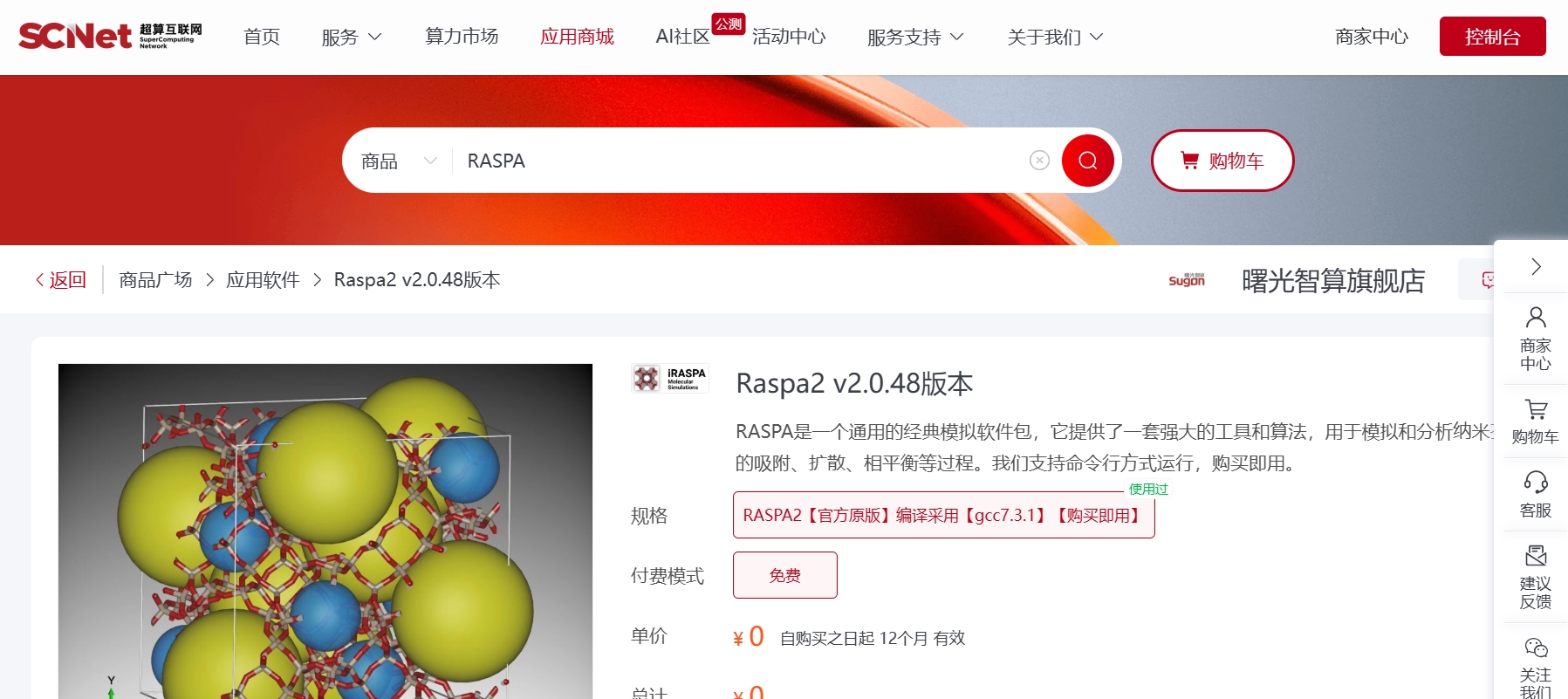
您可在“超算互联网首页”个人账号中找到“我的商品”,选择“应用软件”标签,搜索找到购买的RASPA软件,在右侧选择命令行使用软件。可根据命令行下方的提示信息加载环境变量和使用软件,软件默认安装在家目录的apprepo下,环境变量默认配置在软件包中scripts目录下的env.sh中。在case目录下,有算例文件和用于提交计算任务的slurm脚本。
在商品页面服务与支持有软件的使用手册,接下来我们以RASPA手册中Basic范例的第7项7_Adsorption_of_Methane_in_MFI为例,对计算等温吸附曲线所使用的输入文件进行介绍。输入文件所在位置:
${HOME}/apprepo/raspa2/2.0.48-gcc731/app/examples/Basic/7_Adsorption_of_Methane_in_MFI
进行GCMC模拟所需的文件包括吸附剂坐标文件、吸附质参数文件、力场参数文件和设置参数文件,对simulation.input参数设置文件中的重要内容进行解释:
SimulationType MonteCarlo 模拟的类型是蒙特卡洛模拟
NumberOfCycles 25000 总计进行25000步蒙特卡洛模拟
NumberOfInitializationCycles 5000 初始模拟5000步,即有效采样步为25000-5000=20000步
PrintEvery 1000 每1000步输出一次中间结果
Forcefield ExampleZeolitesForceField 注1
RemoveAtomNumberCodeFromLabel yes
Framework 0 吸附剂编号0,RASPA依照C语言风格,下标从0开始
FrameworkName MFI_SI 吸附剂名称为MFI_SI.cif
UnitCells 2 2 2 构建2*2*2的超晶胞作为吸附剂的初始构象
HeliumVoidFraction 0.29 注2
ExternalTemperature 300.0 模拟温度设置为300K
ExternalPressure 1e4 1e5 模拟压强设置为10000Pa和100000Pa
ComputeNumberOfMoleculesHistogram yes 统计吸附质分子数量并给出直方图
WriteNumberOfMoleculesHistogramEvery 5000
NumberOfMoleculesHistogramSize 1100
NumberOfMoleculesRange 80
ComputeEnergyHistogram yes 统计体系的能量并给出直方图
WriteEnergyHistogramEvery 5000
EnergyHistogramSize 400
EnergyHistogramLowerLimit -110000
EnergyHistogramUpperLimit -20000
Component 0 MoleculeName methane 注3
MoleculeDefinition ExampleDefinitions 吸附质的参数设置文件
TranslationProbability 0.5 吸附质分子的平动几率
ReinsertionProbability 0.5 吸附质分子的再插入几率
SwapProbability 1.0 吸附质分子的交换几率
CreateNumberOfMolecules 0 初始模拟时体系中吸附质分子的数量设置为0个注1:此案例所用力场参数在软件的share文件夹中。力场参数、吸附剂坐标等输入信息,程序的读入顺序是先读share目录,后读当前目录,后读的优先级更高
注2:氦空隙分数表示吸附剂中孔道体积占总体积的百分数,可以使用氦原子的Widom插入模拟来估算这一值,详情参考手册。
注3:此案例所用的吸附质为联合原子模型(united-atom model)的甲烷分子。
更多的关于RASPA功能的参数及设置,可以参考RASPA的官方文档。
提交计算任务
我们以case目录作为当前目录,首先将simulation.input文件复制到当前文件夹,并删掉case文件夹中不相关的文件。复制命令如下:
cp ${HOME}/apprepo/raspa2/2.0.48-gcc731/app/examples/Basic/7_Adsorption_of_Methane_in_MFI/simulation.input .完成操作后文件夹中只保留simulation.input和RASPA2.slurm两个文件。
查看并修改simulation.input文件,我们将温度设置为300K,压强选择10Pa, 100Pa, 200Pa, 400Pa, 800Pa,1000Pa, 5000Pa, 10000Pa, 100000Pa, 1000000Pa, 2000000Pa, 4000000Pa共计12个点。则输入文件需要相应设置为ExternalTemperature 300.0,压强设置写成
ExternalPressure 10 100 200 400 800 1e3 5e3 1e4 1e5 1e6 2e6 4e6修改后保存退出。
接下来检查提交作业脚本RASPA2.slurm,确认队列名称和计算资源,如下列所示。
#!/bin/bash
#SBATCH -J RASPA2 #作业名称
#SBATCH -p kshcsctest #队列名称
#SBATCH -N 1 #节点数量
#SBATCH --ntasks-per-node=1 #每节点核心数
source ${HOME}/apprepo/raspa2/2.0.48-gcc731/scripts/env.sh
export RASPA_DIR=${SCRIPTDIR}/app/install
export DYLD_LIBRARY_PATH=${RASPA_DIR}/lib
export LD_LIBRARY_PATH=${RASPA_DIR}/lib
${RASPA_DIR}/bin/simulate -i simulation.input使用sbatch RASPA2.slurm命令提交计算任务,使用squeue命令查看任务列表,若状态显示R则表示正在计算。
完成计算后,我们需要查看模拟结果,这里我们使用Octave来进行数据可视化,使用VMD v1.9.3查看分子结构。超算互联网提供Octave 8.4.0和VMD v1.9.3版本,支持一键加载环境!购买使用方式可参考上文RASPA的使用流程。
作图之前需要提取出吸附量的数据,我们从case文件夹进入到输出记录的文件夹,使用命令
cd Output/System_0/可以看到相应的软件输出文件,如图所示
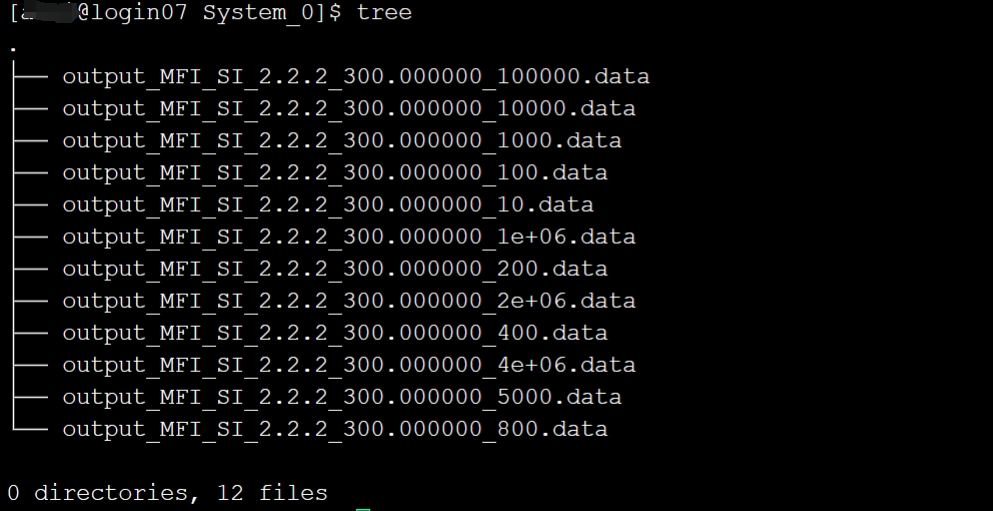
可以使用grep命令将绝对吸附量和过剩吸附量的数据提取出来,命令如下
grep 'Average loading absolute \[mol\/kg' * | awk '{print $1,$7,$9}' >absolute_adsorption.dat
grep 'Average loading excess \[mol\/kg' * | awk '{print $1,$7,$9}' >excess_adsorption.dat获得两个文件,其中absolute_adsorption.dat是绝对吸附量数据,excess_adsorption.dat是过剩吸附量数据。以absolute_adsorption.dat为例,将第一列修改成压强数值,第二列是吸附量(mol/kg),第三列是误差棒。

同理修改excess_adsorption.dat,两个数据文件接下来将用于数据作图。您可以将文件下载至本地,使用自己偏好的软件作图,而本文将展示使用Linux图形桌面的方式进行画图。在使用前,您需要先配置Linux图形桌面。您可在“超算互联网首页”选择右上角的“控制台”,在快捷入口或者图形桌面选项卡中点击“Linux图形桌面”,选择使用软件对应的区域后,“打开桌面”即可打开一个图形桌面,如下图所示。
选择控制台界面的图形管理,找到开启图形桌面的编号,并记录下来,如下图所示。
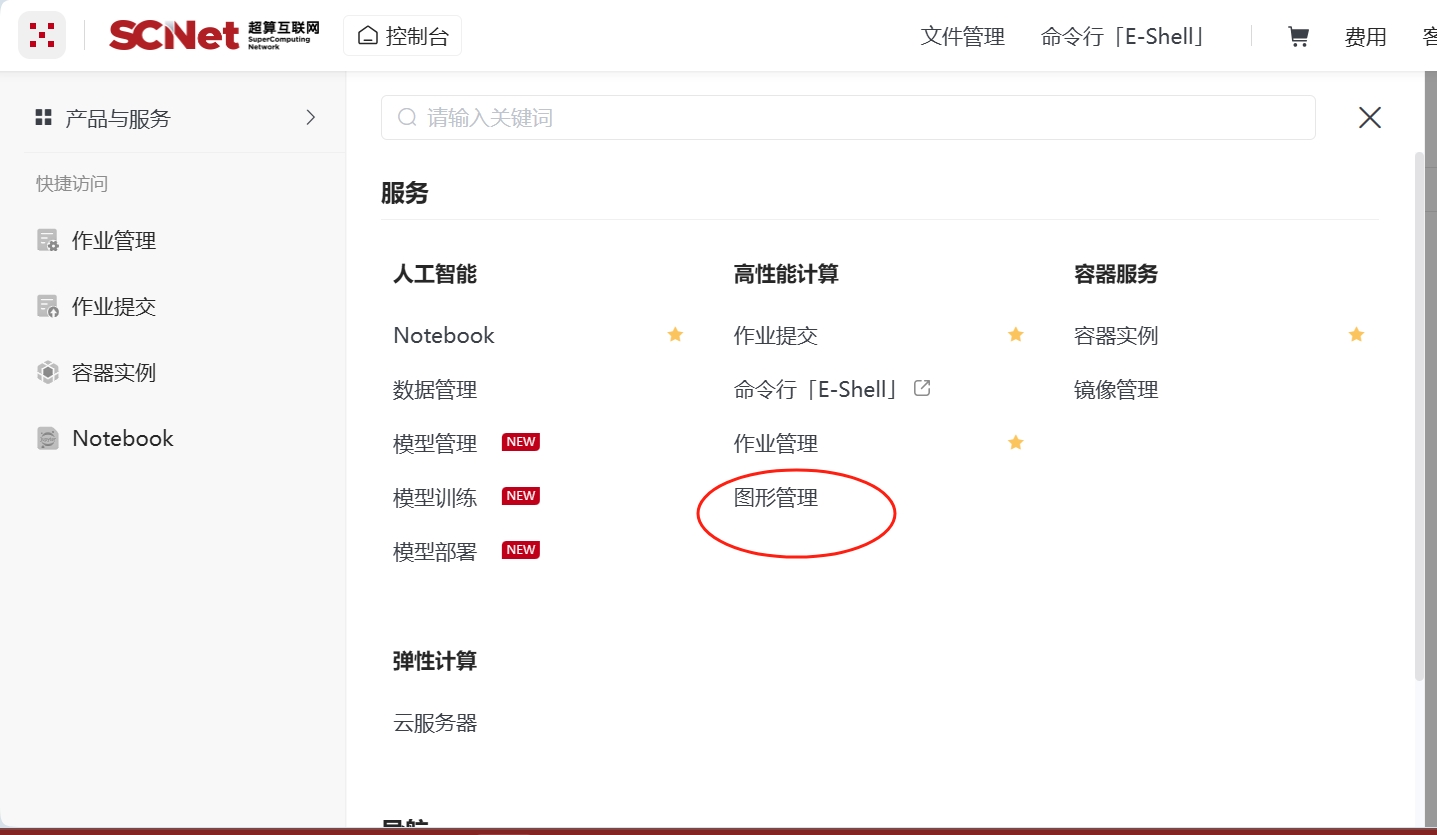
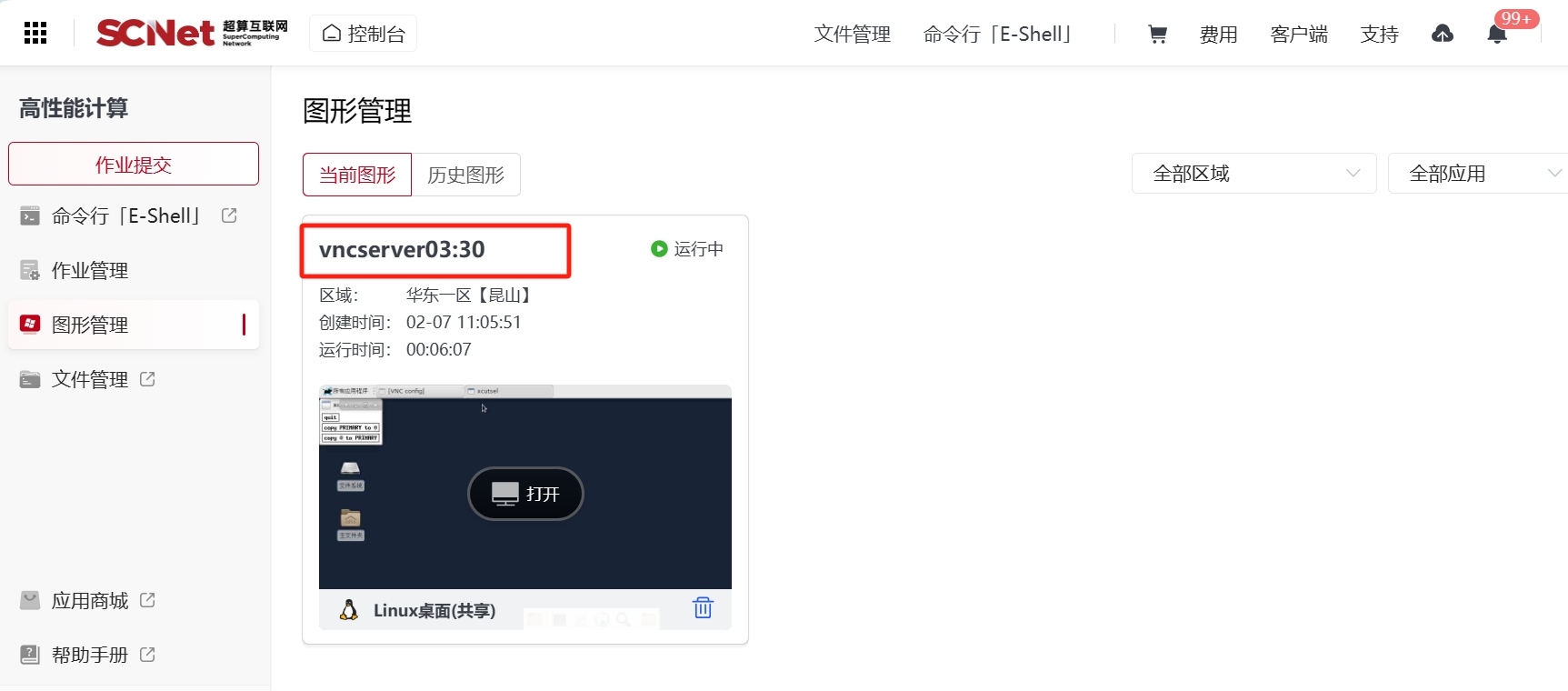
进入Octave的case目录,将absolute_adsorption.dat和absolute_adsorption.dat两个数据文件复制到此文件夹中。回到命令行界面编辑Octave的运行脚本文件octave.slurm,如下所示。注意:export DISPLAY=<端口号>要与上图红色框的内容一致。
#!/bin/bash
module purge
source ${HOME}/apprepo/octave/8.4.0-gnu7.3.1/scripts/env.sh
#需要图形的话,请填写VNC端口号
export DISPLAY=vncserver03:30
octave保存脚本文件后,使用bash octave.slurm启动软件。在Octave提示符后依次输入
A=load('excess_adsorption.dat');
B=sort(A,1);
errorbar(B(:,1),B(:,2),B(:,3));即可在图形化界面中看到等温吸附曲线,如下图所示

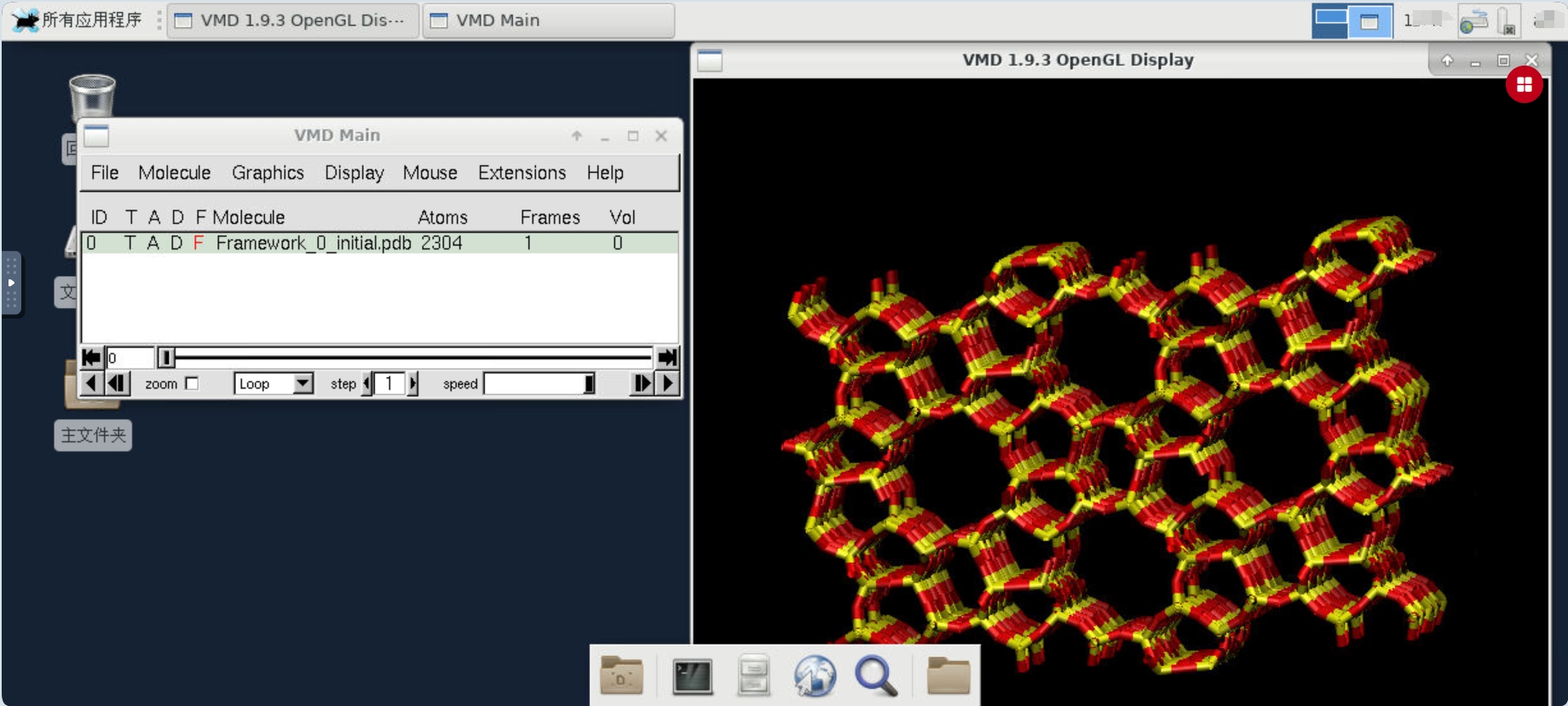
使用VMD查看吸附模型的操作方法与之类似,首先进入VMD的case目录,检查脚本文件,修改端口号。
#!/bin/bash
module purge
source {HOME}/apprepo/vmd/1.9.3-none/scripts/env.sh
#需要修改设置对应的端口号
export DISPLAY=vncserver03:30
vmd运行脚本,可以在桌面环境操作vmd查看吸附体系相应的坐标文件,RASPA输出的坐标文件位于Movies文件夹中。以吸附剂结构为例,可以看到MFI沸石分子筛的空间结构。