高性能计算服务
>
软件专区
>
最佳实践
>
ABACUS实操:从零构建千级原子规模进行SCF计算
在材料科学和量子化学领域,第一性原理计算是一个强大的工具,使研究人员能够从理论层面预测和分析材料的性质。作为国内自主研发的开源第一性原理计算软件,ABACUS(原子算筹)允许研究人员根据需求采用平面波基组或数值原子轨道基组进行电子自洽迭代(SCF)、能带、态密度计算、结构优化、分子动力学模拟等多种计算。
本次实操,我们将从千级规模的原子构建开始,逐步讲解如何在超算互联网使用ABACUS完成SCF计算。
ABACUS进行SCF计算时,一般需要STRU、KPT和INPUT这三个基本输入文件。
作为ABACUS程序运行的必要输入之一,STRU文件提供了晶格矢量、元素种类、原子位置、数值原子轨道、赝势文件等基本的结构相关的信息,是ABACUS软件中进行材料模拟和计算物理研究的基础。本节我们将以Si原子为例,讲解如何构建STRU文件。
首先需要有一个含有Si原子的初始CIF结构文件,我们选择从Materials Project官网上下载一个初始的结构。Materials Project是一个强大的资源,为材料研究人员提供了设计更好材料所需的信息,通过调用官方提供的API,我们编写如下Python脚本直接下载选择的“material id”。
from mp_api.client import MPRester
from pymatgen.io.cif import CifWriter
with MPRester(api_key="Your_API_KEY_From_MP_website") as mpr:
structure = mpr.get_structure_by_material_id("mp-1201492")
cif_writer = CifWriter(structure)
cif_writer.write_file("Si-232.cif")这里我们选择id号为mp-1201492的结构,含有232个Si原子,并将结构文件命名为Si-232.cif。使用脚本前,请替换您在Materials Project的API KEY和想要下载的结构id。CIF结构文件可以使用VESTA软件直接可视化展示,您可以使用超算互联网的VESTA图形软件:  VESTA软件:https://www.scnet.cn/ui/mall/detail/goods?type=software&common1=APP_SOFTWARE&id=1815649843946000386&resource=APP_SOFTWARE&keyword=VESTA 该软件支持命令行和图形两种运行方式,购买即用。我们使用图形的方式运行VESTA,加载下载的Si-232.cif文件,展示如下。
VESTA软件:https://www.scnet.cn/ui/mall/detail/goods?type=software&common1=APP_SOFTWARE&id=1815649843946000386&resource=APP_SOFTWARE&keyword=VESTA 该软件支持命令行和图形两种运行方式,购买即用。我们使用图形的方式运行VESTA,加载下载的Si-232.cif文件,展示如下。 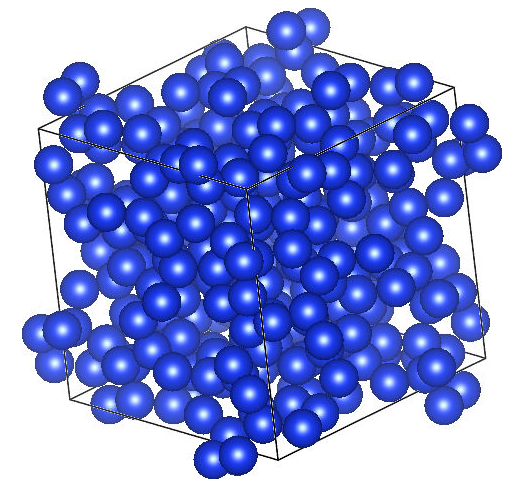 接下来拓展已有的原子规模使其达到千级,这里使用ASE库来扩展原子规模,Python脚本参考如下:
接下来拓展已有的原子规模使其达到千级,这里使用ASE库来扩展原子规模,Python脚本参考如下:
from ase.io import read, write
from ase import Atoms
# 读取cif文件
atoms = read('Si-232.cif')
# 原始晶胞中有232个原子,我们在x、y、z方向分别扩展3、2、1倍,这样就会有1392个si原子
x_ext = 3
y_ext = 2
z_ext = 1
# 创建超胞
supercell = atoms * (x_ext, y_ext, z_ext)
# 保存为新的cif文件
write('Si-1392.cif', supercell)Atomic Simulation Environment (ASE) 是一个用于设置、操作、运行、可视化和分析原子模拟的工具和Python模块集合,非常适合计算化学和计算材料领域的研究人员使用且容易上手。在使用前您需要在超算互联网安装加载python3环境,推荐安装链接如下:  加载python3环境后,通过“pip install ase”来安装ase模块。通过python运行上述脚本,我们将232个Si原子扩展到了1392个Si原子,同样使用VESTA可视化结构如下。
加载python3环境后,通过“pip install ase”来安装ase模块。通过python运行上述脚本,我们将232个Si原子扩展到了1392个Si原子,同样使用VESTA可视化结构如下。 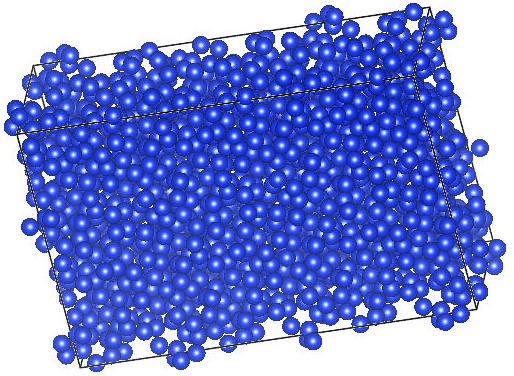 通过前两步,我们生成了千级原子规模的CIF结构文件,接下来就需要将CIF格式的文件转换成ABACUS的STRU格式文件。有两种方法可以将CIF文件转换成STRU文件,一种是使用ABACUS开发者写好的ASE-ABACUS接口。此种方法您需要安装具有abacus接口的ase,而不是直接安装ase库,参考脚本如下:
通过前两步,我们生成了千级原子规模的CIF结构文件,接下来就需要将CIF格式的文件转换成ABACUS的STRU格式文件。有两种方法可以将CIF文件转换成STRU文件,一种是使用ABACUS开发者写好的ASE-ABACUS接口。此种方法您需要安装具有abacus接口的ase,而不是直接安装ase库,参考脚本如下:
git clone https://gitlab.com/1041176461/ase-abacus.git
cd ase-abacus
pip install .转换代码您可以参考ABACUS官网,我们这里采用第二种方式,即使用ATOMKIT跨平台建模软件,使用其101功能可以快速完成结构转化。超算互联网同样也预装了ATOMKIT软件,您只需简单的购买操作即可以命令行方式使用该软件,软件如下:  Atomkit软件:https://www.scnet.cn/ui/mall/detail/goods?type=software&common1=APP_SOFTWARE&id=1773262475209510914&resource=APP_SOFTWARE&keyword=atomkit 命令行启动atomkit软件,接着输入101 -->回车,输入113 Si-1392.cif-->回车,即可生成Si-1392.STRU,在随后的运行中,我们将其命名为STRU。
Atomkit软件:https://www.scnet.cn/ui/mall/detail/goods?type=software&common1=APP_SOFTWARE&id=1773262475209510914&resource=APP_SOFTWARE&keyword=atomkit 命令行启动atomkit软件,接着输入101 -->回车,输入113 Si-1392.cif-->回车,即可生成Si-1392.STRU,在随后的运行中,我们将其命名为STRU。 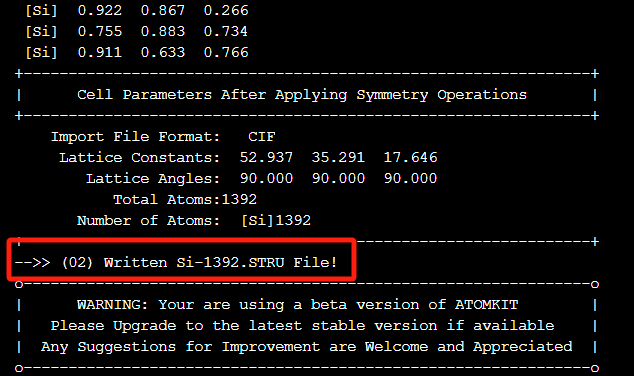 我们来分析并调整生成的STRU文件。下面是STRU文件的主要信息:
我们来分析并调整生成的STRU文件。下面是STRU文件的主要信息:
ATOMIC_SPECIES
Si 28.085 Si_ONCV_PBE-1.0.upf
NUMERICAL_ORBITAL
Si_gga_7au_100Ry_2s2p1d.orb
LATTICE_CONSTANT
1.889726
LATTICE_VECTORS
52.93653063000 0.000000000000 0.000000000000
0.000000000000 35.29102042000 0.000000000000
0.000000000000 0.000000000000 17.64551021000
ATOMIC_POSITIONS
Direct
Si
0.000
1392
0.087506863333 0.422953050000 0.418132640000 1 1 1 mag 0.0
0.245826470000 0.172953050000 0.081867360000 1 1 1 mag 0.0
0.079159803333 0.077046950000 0.918132640000 1 1 1 mag 0.0
0.254173530000 0.327046950000 0.581867360000 1 1 1 mag 0.0“ATOMIC_SPECIES”提供了元素种类、原子质量,并指定了使用的赝势文件为Si_ONCV_PBE-1.0.upf;“NUMERICAL_ORBITAL”指定了Si原子的数值轨道文件,该文件包含了计算所需的波函数信息;“LATTICE_VECTORS”定义晶体的晶格向量,它们用于描述晶体的尺寸和形状。“ATOMIC_POSITIONS”定义了原子在晶体中的位置,Direct表示原子位置是以晶格坐标系给出的。文件里提到了另外两个文件:upf和orb,这里我们可以修改想要的赝势和数值轨道文件,同时,abacus官方软件包的PP_ORB文件夹内也提供了相应的文件,我们实验过程中修改并使用其中的Si_ONCV_PBE-1.0.upf和Si_gga_6au_100Ry_2s2p1d.orb。
至此,我们就构建出了含有1392个Si原子的STRU文件,也完成了SCF计算的结构信息文件生成。
第二步我们开始构建KPT文件。KPT文件提供周期性边界条件下布里渊区k点采样的网格设置,这些点在计算晶体的电子结构时非常重要。我们使用如下内容:
K_POINTS
0
Gamma
1 1 1 0 0 0K_POINTS是指开始定义K点的部分,第2行表示k点的总数,设置成“0”代表 K点自动生成,第三行的“Gamma”指明使用Gamma点的计算,代表布里渊区的中心。最后有六个整数,前三个代表网格沿着x、y、z三个方向换分成1份,后三个表示网格不平移。整个KPT文件表示我们采用1个Gamma点作为K点。 K点数与计算规模和计算精度之间的关系非常密切,增加K点数可以提高计算精度,因为更多的K点可以更好地采样布里渊区,同时计算的时间也会越长,内存消耗也会更大。考虑到我们模拟的原子规模比较大,而且本文暂不考虑k点对收敛性等的影响,所以做了如上简单的设置。
在ABACUS中,INPUT文件提供了计算所需的各种参数设置。输入文件的设置直接影响到计算的成功与否以及结果的准确性,有时也需要进行多次试验来调整参数以达到最佳效果,结合我们将要进行的SCF计算,主要包括以下几个重要部分:
1.全局设置:也可以叫基本参数设置,通常包括对计算的基本设置,如计算类型(比如优化、能带计算等)、赝势文件目录、计算的基组设置等一些全局参数。比如本文进行scf计算,所以calculation设置为‘scf’。
2.SCF迭代参数:这里我们主要考虑2个参数,分别是scf_nmax和scf_thr。前者用来设置scf电子迭代的最大次数,后者代表两个相邻电子迭代步之间的电荷密度误差,是SCF计算中判断是否收敛、完成计算的标准。
3.KS方程求解:ks_solver是ABACUS软件中用于计算 Kohn-Sham 哈密顿矩阵对角化的不同方法。前述全局设置中,我们设置计算的基组“basis_type”为pw,即平面波基组,所以有cg(共轭梯度法)、bpcg(双共轭梯度法)和dav(Davidson)等算法可供选择。cg算法是默认方法,具有较好的收敛性和效率,我们采用该算法。另外一个参数是nbands,即计算KS轨道数目。本文主要目的是介绍SCF计算,暂不考虑收敛性和精度等,这里采用默认的nbands数。
下面是我们使用的主要INPUT参数,更多关于INPUT的参数解释请参考ABACUS官网: https://abacus.deepmodeling.com/en/latest/advanced/input_files/input-main.html
INPUT_PARAMETERS
#Parameters (1.General)
suffix Si
calculation scf
symmetry 1
pseudo_dir .
orbital_dir .
basis_type pw
ecutwfc 100
#Parameters (2. SCF iterations)
scf_nmax 100
scf_thr 1e-8
#
#Parameters (3. Solve KS equation)
#bands default:max(1.2*occupied_bands, occupied_bands + 10)
ks_solver cg
#Parameters (4.Smearing)
smearing_method gauss
smearing_sigma 0.01经过前面的工作,我们已经准备好运行scf计算必要的STRU、KPT和INPUT文件,把赝势文件和数值轨道文件拷贝到当前目录(在INPUT文件中设置了这两个文件在当前目录下),接下来就可以准备运行ABACUS执行SCF计算了。我们使用slurm提交作业,具体脚本内容如下:
#!/bin/bash
#SBATCH -J aba-Si-1392
#SBATCH -p tyhcnormal
#SBATCH -N 16
#SBATCH --ntasks-per-node=8
#SBATCH --exclusive
#SBATCH -c 8
module purge
source /PATH/TO/YOUR/HOME/apprepo/abacus/v3.6.1-intelmpi2021/scripts/env.sh
export OMP_NUM_THREADS=8
mpirun -np $SLURM_NTASKS abacus我们使用128进程和8线程来运行我们的计算,使用sbatch命令提交作业,由于原子数量较大,运行时间较长,部分输出截图如下: 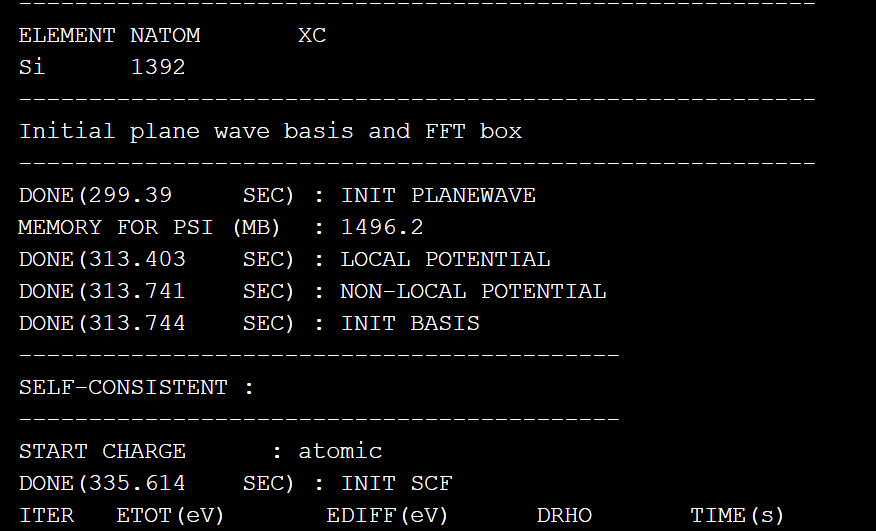 本篇文章,我们从头讲述了如何构建必要文件并运行ABACUS进行scf计算,手把手介绍了如何使用ASE库、Atomkit等配套软件来构建和转化STRU文件,如何设置KPT和INPUT文件参数并成功运行ABACUS软件。
本篇文章,我们从头讲述了如何构建必要文件并运行ABACUS进行scf计算,手把手介绍了如何使用ASE库、Atomkit等配套软件来构建和转化STRU文件,如何设置KPT和INPUT文件参数并成功运行ABACUS软件。
希望本篇文章能够给您在使用ABACUS上有一定的借鉴意义。