高性能计算服务
>
软件专区
>
最佳实践
>
如何使用ORCA进行单点能计算、结构优化、计算IRRaman振动光谱
ORCA是一个灵活高效易用的量子化学计算工具,着重于开壳层分子的光谱学性质计算。
ORCA包含了一系列的标准化量子化学计算方法,从半经验方法到DFT,以及单参考和多参考从头算方法,还可以处理环境效应和相对论效应。
本次实操,我们详细介绍如何使用ORCA进行单点能计算、结构优化、计算IR/Raman振动光谱,通过清晰的操作步骤、输入文件示例(以对甲苯酚为模型分子)以及频率分析,帮助科研人员深入掌握ORCA的核心计算流程。
红外(IR)光谱又称分子振动转动光谱,属于分子吸收光谱。样品受到频率连续变化的红外光照射时,分子吸收其中一些频率的辐射,分子振动或转动引起偶极矩的净变化,使振动-转动能级由基态跃迁到激发态,相应于这些区域的透射光强减弱,记录百分透过率T%对波数或波长的曲线,即红外光谱。红外光谱主要用于化合物鉴定及分子结构表征,亦可用于定量分析。红外光谱可分为近红外区、中红外区和远红外区。其中4000~400 cm⁻¹ 的中红外区是红外光谱的主要分析区域,其吸收带主要是绝大多数有机化合物和无机离子的基频吸收产生,适合进行定性分析。
拉曼(Raman)光谱是一种基于拉曼散射效应的光谱分析技术,用于研究分子的振动、转动等低能级跃迁,从而提供分子结构、化学组成等信息。当一束单色光照射到样品时,大部分光子会发生弹性散射(瑞利散射),散射光的频率与入射光相同,小部分光子会发生非弹性散射,即拉曼散射,散射光的频率相对于入射光发生了变化。散射光频率与入射光频率之差称为拉曼位移,其与入射光频率无关,与样品的分子振动或转动能级有关。拉曼光谱图的横轴为拉曼位移,代表分子振动/转动模式的特征频率,纵轴是散射光强度,反映特定振动模式发生的概率,谱峰位置对应特定化学键或官能团的振动模式,谱峰强度/形状反映分子浓度、对称性等信息。
二者的区别:拉曼光谱依赖分子的极化率变化(对称振动敏感);红外光谱依赖分子偶极矩变化(不对称振动敏感)
注:本次最佳实践主要参考了ORCA手册6.12.3节IR/Raman Spectra, Vibrational Modes and Isotope Shifts的内容
我们将以对甲苯酚分子为例,展示如何使用ORCA量子化学计算软件获得分子性质。首先我们使用Avogadro软件绘制出对甲苯酚的分子结构,如图1所示。 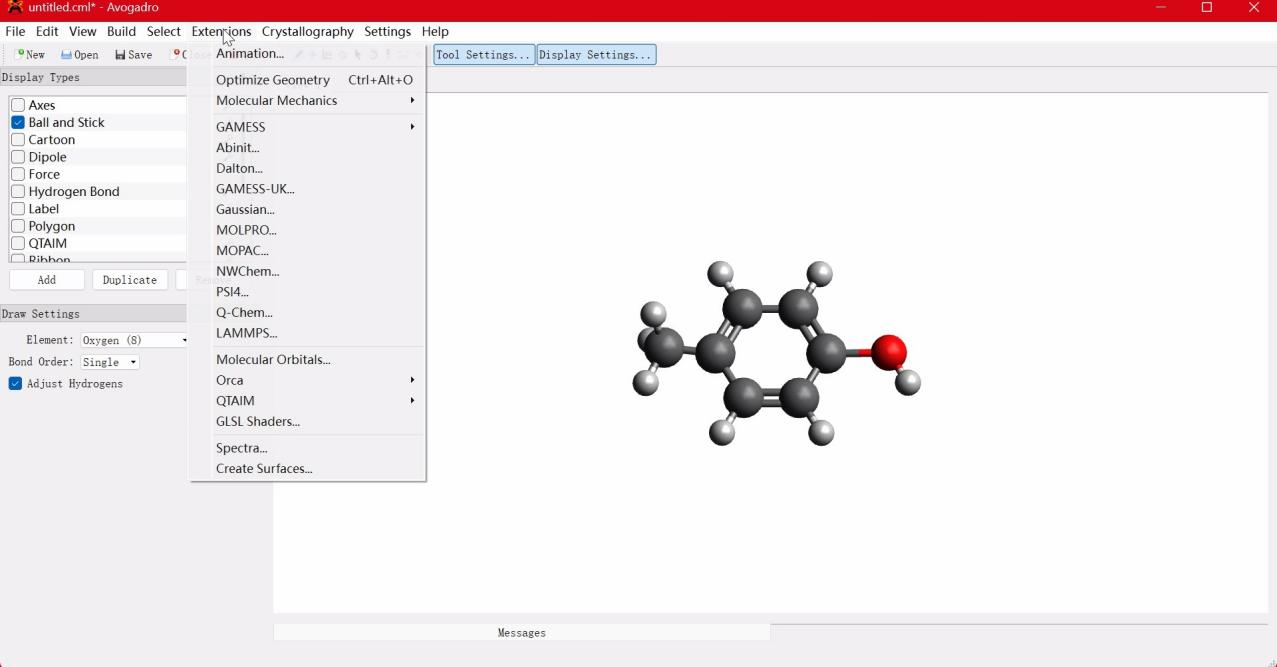 图1 使用Avogadro软件生成输入文件
图1 使用Avogadro软件生成输入文件
在超算互联网的应用商城中点击使用ORCA 6.0.1(图2),即可在控制台进入使用界面。 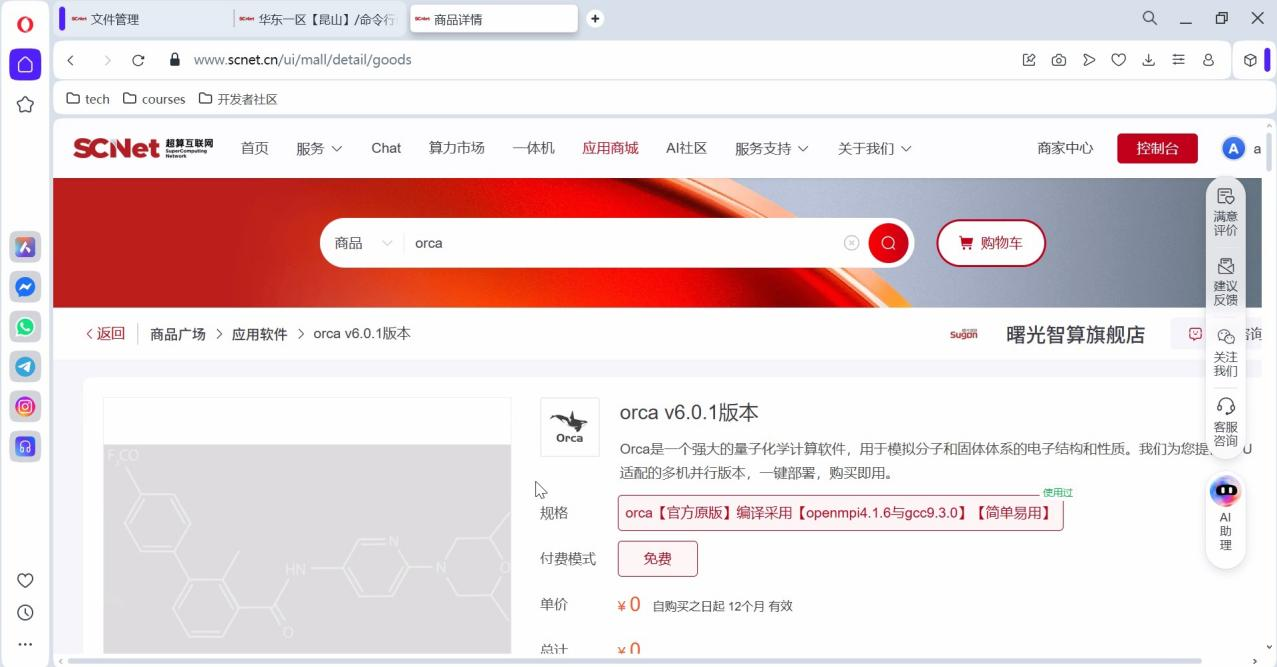 图2 应用商城中的ORCA应用下单界面
图2 应用商城中的ORCA应用下单界面
检查软件目录,在case目录中可以看到示例输入文件和提交任务的脚本orca.slurm ,根据需要修改提交任务脚本中所需要的节点数和核数,并将输入文件替换成刚上传的对甲苯酚的输入文件名。检查ORCA输入文件,此处我们选用B3LYP方法,def2-SVP基组,用户可以查阅ORCA软件手册,根据需要选择合适的方法和基组。关键词SP 表示单点能计算。
! B3LYP SP def2-SVP
* xyz 0 1
H 1.27806 -0.15174 2.14021
C 0.76594 -0.08132 1.18369
C -0.62648 -0.02337 1.14962
H -1.18563 -0.04922 2.08190
C -1.29796 0.06767 -0.07181
C -0.56323 0.09934 -1.25846
H -1.07476 0.16997 -2.21551
C 0.82895 0.04132 -1.22531
H 1.39445 0.06687 -2.15389
C 1.49505 -0.04860 -0.00449
C -2.81445 0.13321 -0.11911
H -3.24569 0.08948 0.88362
H -3.21314 -0.70285 -0.69813
H -3.13972 1.06387 -0.58954
O 2.85664 -0.10352 0.02152
H 3.16970 -0.15886 0.94199
*使用sbatch orca.slurm即可提交计算任务,查看输出文件可以得到单点能数据
FINAL SINGLE POINT ENERGY -346.322735994173
注意:根据所选择的方法和基组的不同,计算得到的单点能数据会有不同。
接下来修改输入文件,将输入文件中的关键词SP 替换成OPT ,则计算任务变成了结构优化。计算任务结束,我们将得到优化过的分子结构,可以使用文件管理下载,在Avogadro软件中打开查看。
红外光谱
使用ORCA进行频率分析计算时会自动计算红外光谱的强度,输出内容如下列所示,其中第一列“Mode”表示振动模式,第二列是具体的波数,第三列是摩尔吸收系数ε,这个值与红外光谱的强度有关。
-----------
IR SPECTRUM
-----------
Mode freq eps Int T² TX TY TZ
cm⁻¹ L/(mol*cm) km/mol a.u.
----------------------------------------------------------------------------
6: 27.90 0.000090 0.45 0.001006 (-0.001233 -0.031615 -0.002232)
7: 147.85 0.000198 1.00 0.000419 ( 0.000864 0.020426 0.001003)
8: 308.08 0.000376 1.90 0.000381 (-0.017887 0.000618 0.007768)
9: 336.22 0.000034 0.17 0.000031 (-0.000164 -0.005568 -0.000267)
10: 373.22 0.021557 108.94 0.018025 (-0.005193 -0.133982 -0.006834)
11: 427.46 0.000047 0.24 0.000034 ( 0.001363 0.005687 0.000459)
12: 429.58 0.001844 9.32 0.001340 (-0.036096 0.001726 -0.005821)
...接下来为了绘制红外光谱,我们可以使用ORCA软件自带的工具,具体的命令是
orca_mapspc freq.out ir -w25
orca_mapspc命令的基础用法包括
输入命令后,就可以得到相应的数据文件freq.out.ir.dat,即可以放在数据作图软件中绘制红外光谱图。可以根据需要选择作图软件,作图效果如图3所示。
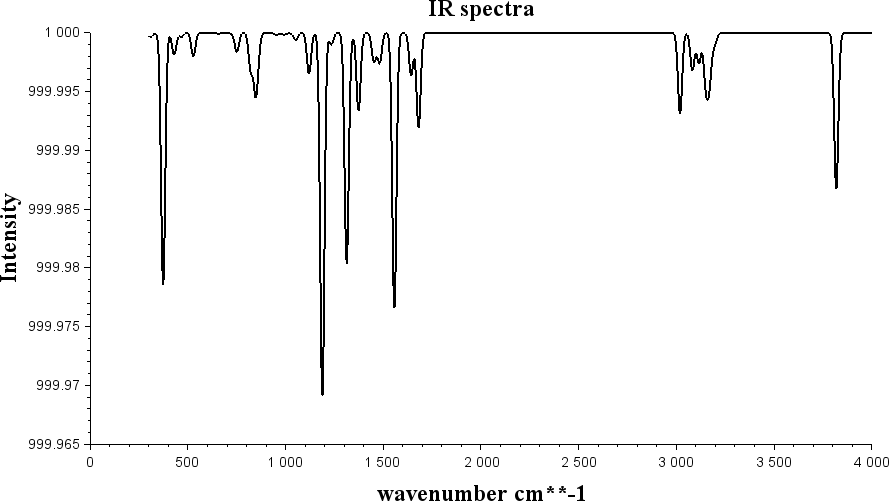 图3:红外振动光谱
图3:红外振动光谱
为了查看每一个振动频率对应的振动模式,我们可以使用orca_pltvib工具将振动模式对应的坐标文件导出,并放入Avogadro软件中观察。操作如下
orca_pltvib freq.out all输出的振动模式的编号与freq.out输出文件中的振动模式编号相匹配。
注:对于近红外区的倍频和组合频带计算,以及VPT2校正请参考手册。
拉曼光谱
为了计算拉曼光谱,我们需要依据简正模式去计算极化率的导数,如果我们使用了数值法的频率计算(即关键词使用了NumFreq)并且还进行极化率的计算,那么程序会自动计算其拉曼光谱。例如我们将输入文件修改成如下内容:
! B3LYP OPT NumFreq def2-SVP TightSCF
%elprop Polar 1 end
* xyz 0 1
H 1.28036 -0.15251 2.16123
C 0.76720 -0.08115 1.19610
C -0.62990 -0.02246 1.14891
H -1.19237 -0.04886 2.08713
C -1.31997 0.06873 -0.06703
C -0.55167 0.09917 -1.24561
H -1.05723 0.17008 -2.21398
C 0.83980 0.04178 -1.21694
H 1.42733 0.06615 -2.13750
C 1.51256 -0.04928 0.01155
C -2.82802 0.13326 -0.12238
H -3.27077 0.09194 0.88439
H -3.24845 -0.70368 -0.70600
H -3.17647 1.06428 -0.60170
O 2.87195 -0.10294 -0.00881
H 3.20338 -0.16230 0.89693
*修改并保存,提交计算任务。 计算任务完成后,可以在输出文件中查看相应的拉曼振动的信息。
--------------
RAMAN SPECTRUM
--------------
Mode freq (cm⁻¹) Activity Depolarization
-------------------------------------------------------------------
6: 46.49 0.379499 0.749981
7: 146.89 0.035869 0.718853
8: 300.87 0.239859 0.422940
9: 333.55 1.700708 0.747390
10: 362.37 4.123520 0.742487
11: 422.24 0.005112 0.749008
12: 425.44 0.104724 0.740295
...使用orca_mapspc命令可以获得拉曼光谱数据,即 orca_mapspc raman.out raman -w50 将得到的后缀为*.raman.out.dat数据文件作图效果如图4所示。 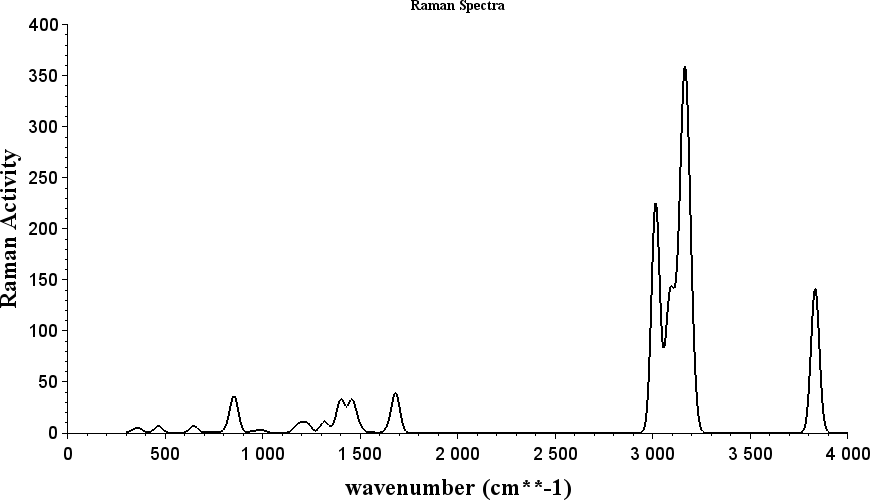 图4:拉曼光谱
图4:拉曼光谱