高性能计算服务
>
软件专区
>
最佳实践
>
ABACUS-DeePMD-LAMMPS驱动的高效、高精度分子动力学模拟
分子动力学(MD)模拟广泛应用于物理学、化学、材料科学、生物医药等科学和技术领域。
在介绍主要模拟过程之前,我们先了解几个概念:
①分子动力学(Molecular Dynamics,MD):利用数值方法求解牛顿运动方程来研究体系随时间演化的计算方法,广泛用于研究与原子运动路径相关的一些过程,如扩散、相变、化学反应等。
②从头算分子动力学(Ab Initio Molecular Dynamics,AIMD):采用第一性原理方法如密度泛函理论(DFT)来计算体系的势能面,又称为第一性原理的分子动力学方法(FPMD),特点是精度高,但计算量巨大,计算昂贵。
③经典分子动力学(Classical Molecular Dynamics,CMD):通过构建描述原子间相互作用的势函数来获得每个原子的受力情况,进而利用积分运动方程来获得每一个原子下一个时刻的空间位置,从而获得随时间演化的粒子的速度和位置,特点是计算量较小,但精度差。
④深度势能分子动力学(Deep Potential Molecular Dynamics,DPMD):基于深度学习(Deep Learning)技术的分子动力学模拟方法,实现高效模拟分子间复杂作用。相比传统的MD,DPMD无需手动构建势函数,而是通过深度神经网络来学习分子间的相互作用势能函数,特点是确保高计算精度的同时计算量较小,能够兼顾AIMD和CMD的优点。
为实现高效、高精度的分子动力学模拟,可采用 “DFT-DPMD-MD”模拟路线:使用三款分子动力学模拟软件(ABACUS、DeePMD、LAMMPS),借助 DeepMD 软件,对基于第一性原理的 ABACUS 分子动力学软件模拟数据进行 DP 模型训练,然后运用兼容DP模型的经典分子动力学模拟软件LAMMPS进行快速计算。
本次实操,我们详细介绍了如何在超算互联网平台申请软硬件资源,使用三款分子动力学模拟软件对氯化锂(LiCl)的局部结构和输运特性进行模拟,并且对模拟结果进行可视化处理,从而实现“DFT-DPMD-MD”分子动力学模拟路线,助力材料科学领域研究。
本次计算需要用到三款分子动力学软件。
1、ABACUS
ABACUS(Atomic-orbital Based Ab-initio Computation at UStc)是一款基于第一性原理的国产开源分子动力学软件,主要用于大规模电子结构计算、原子结构优化及分子动力学模拟。目前超算互联网平台已经部署各种版本的ABACUS。
2、DeePMD
DeePMD(Deep Potential Model)是一款基于深度学习的分子动力学模拟软件,主要用于预测原子间相互作用力,从而加速和改进分子动力学模拟的精度和效率。软件通过学习大量第一性原理数据训练高精度的DP模型,以准确预测原子的相互作用。目前超算互联网平台已经部署各种版本的DeePMD。
3、LAMMPS
LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一款开源的经典分子动力学模拟软件,可以模拟气态、液态或固态的粒子系统。软件可以计算几个粒子到上百万甚至上亿粒子的体系。目前超算互联网平台已经部署各种版本的Lammps。
本次模拟需要提前配置相应的软硬件资源,包括:
1、硬件
三款软件均支持异构计算,您可以在算力市场申请使用相应配置的算力资源,本例使用的是“曙光智算旗舰店-异构加速卡1 显存16GB-华东一区【昆山】”。 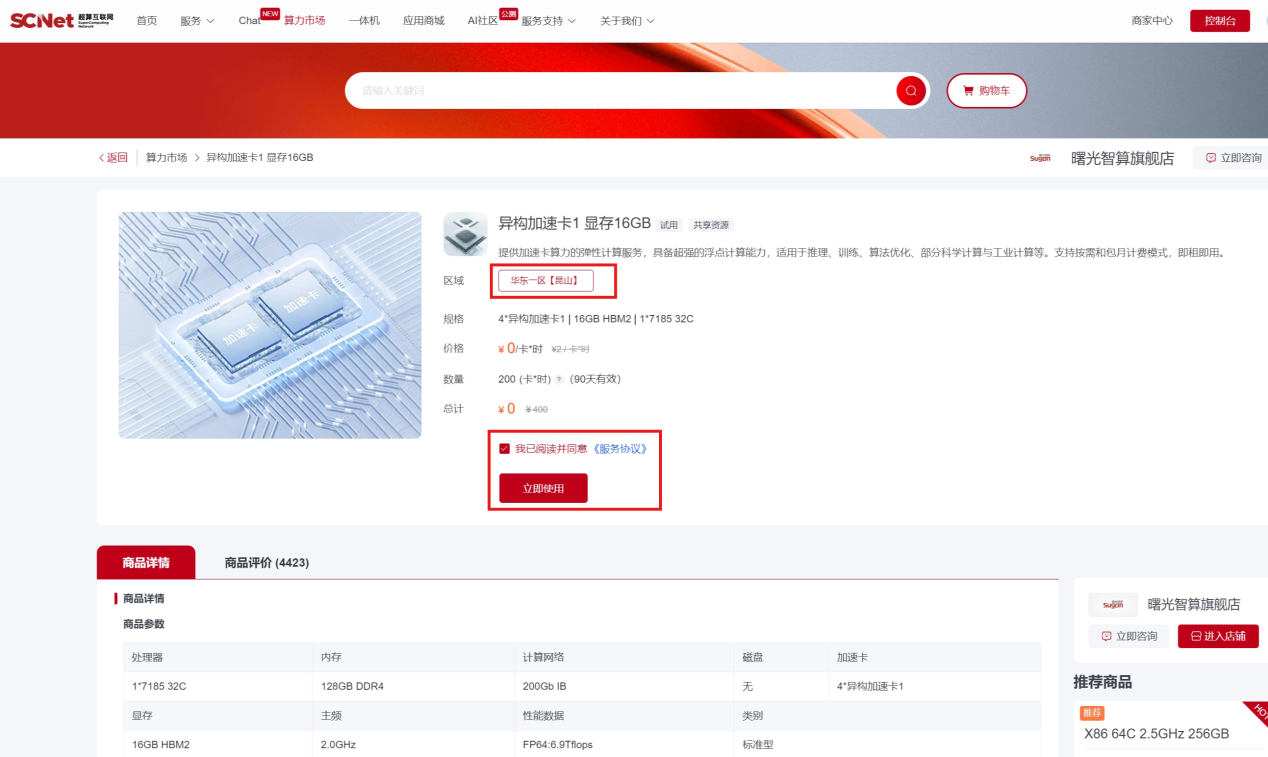 如下图所示,您可以按照Step1→Step3的顺序,通过点击“我的资源”→“高性能计算”查看申请的算力资源。
如下图所示,您可以按照Step1→Step3的顺序,通过点击“我的资源”→“高性能计算”查看申请的算力资源。  2、软件
2、软件
① ABACUS v3.6.0加速版
超算互联网平台提供预编译的ABACUS v3.6.0加速版软件,可以直接申请使用。
② Deepmd
超算互联网平台提供预编译的Deepmd v3.0.0b4加速版软件,可以直接申请使用。您可以通过以下链接直接购买使用,这里使用的是dtk23.10编译的tensorflow2.11.0加速版。
③ Lammps
超算互联网平台提供预编译的Lammps 2Aug2023版软件,可以直接申请使用。您可以通过以下链接直接购买使用,这里使用的是gcc7.3.1与hpcx编译的版本。
在模拟前,我们可以在家目录使用“mkdir LiCl-test”命令新建测试文件夹LiCl-test,进入该文件夹后再分别新建四个文件夹“abacus-data”、“deepmd-data”、“lammps-data”和“lammps-md-data”,对应本次使用的三款软件,用于存放对应的数据文件。如下图所示。 
cd进入abacus-data文件夹,将ABACUS计算所需的算例文件上传,包括INPUT文件、STRU文件、KPT文件、.upf文件和.orb文件。各文件作用为: 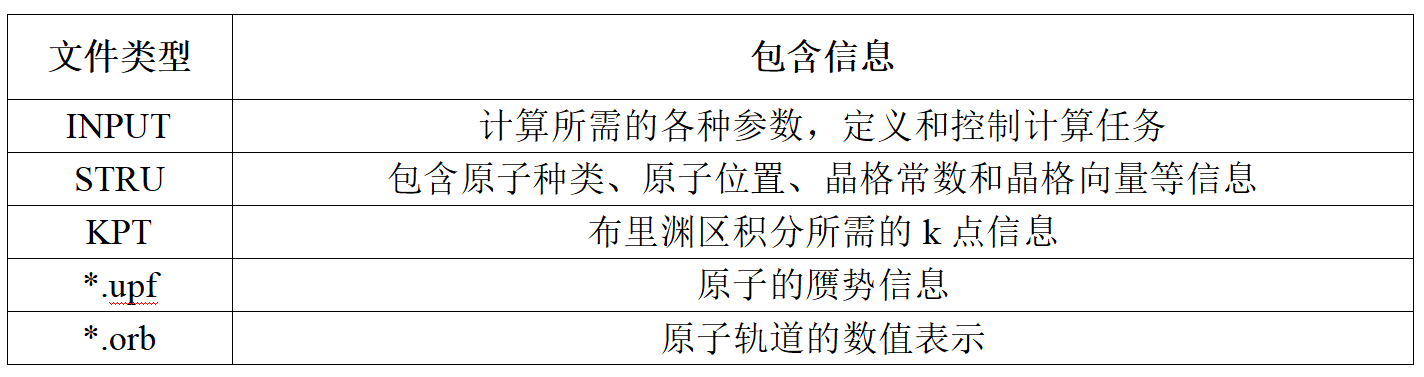
软件申请成功后,默认安装在家目录的apprepo下,环境变量默认放在软件scripts目录下的env.sh中,通过修改case文件夹的脚本文件abacus.slurm得到我们需要的脚本文件,如下图所示。我们使用单个节点的4个CPU核和4张异构加速卡AI进行计算,单核8线程。 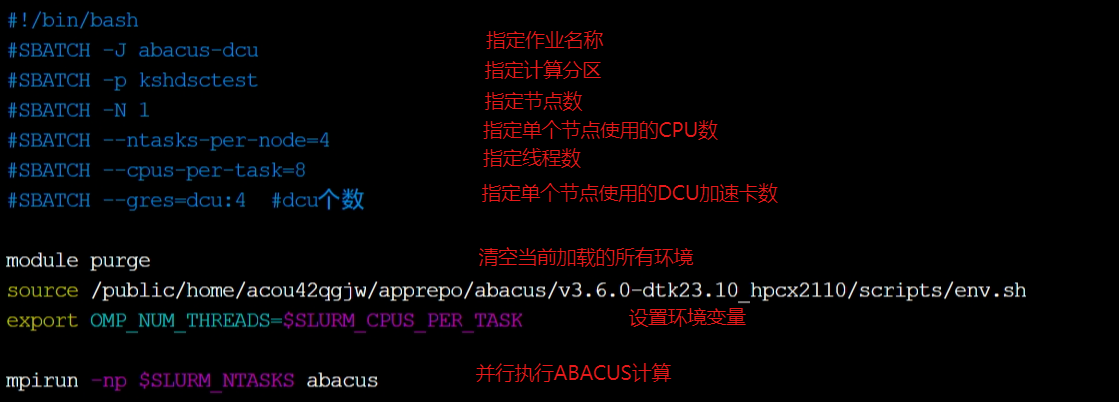 使用sbatch abacus.slurm提交计算任务;使用squeue查看作业情况;使用tail -f slurm-***.out动态查看软件运行输出。如下图所示。
使用sbatch abacus.slurm提交计算任务;使用squeue查看作业情况;使用tail -f slurm-***.out动态查看软件运行输出。如下图所示。 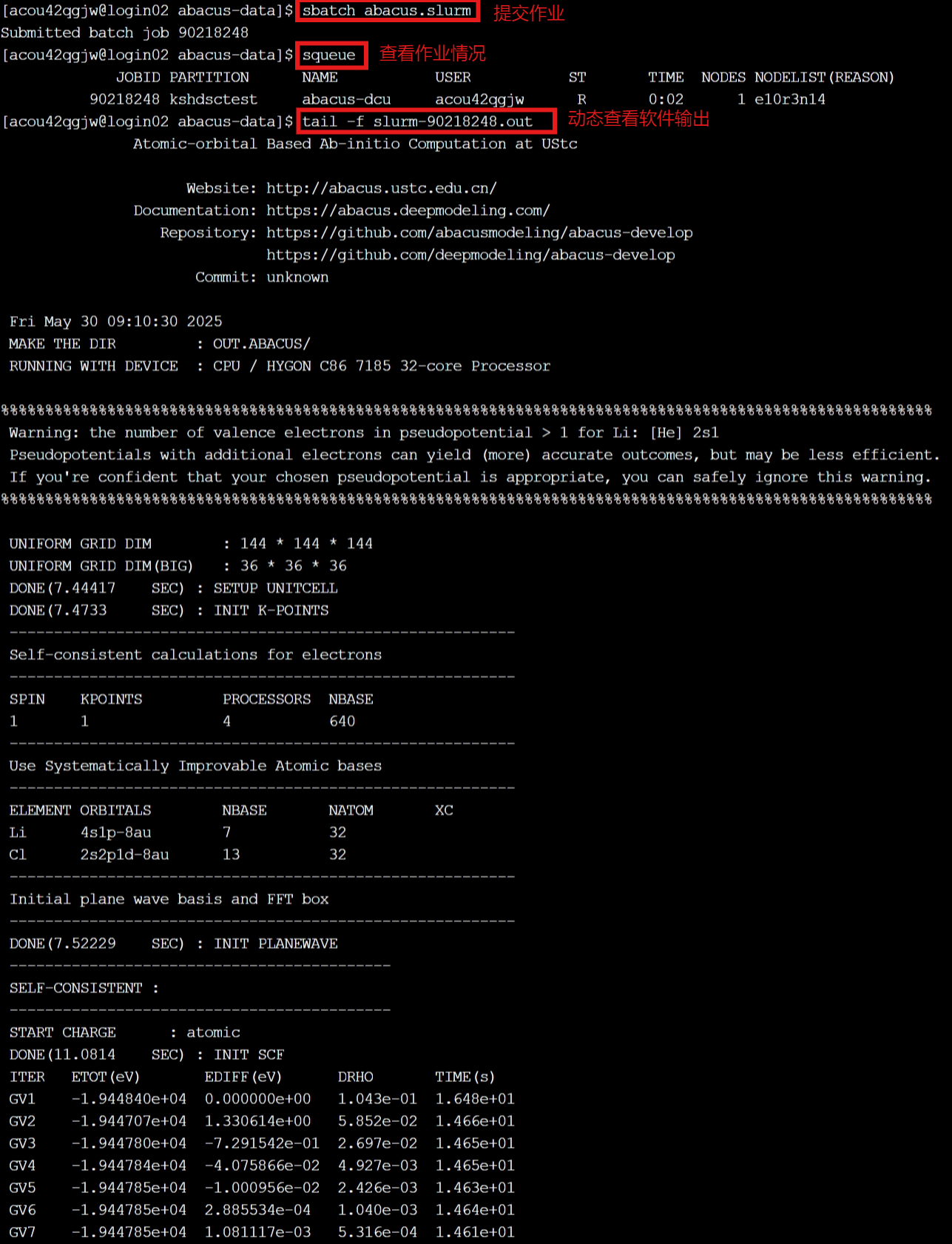
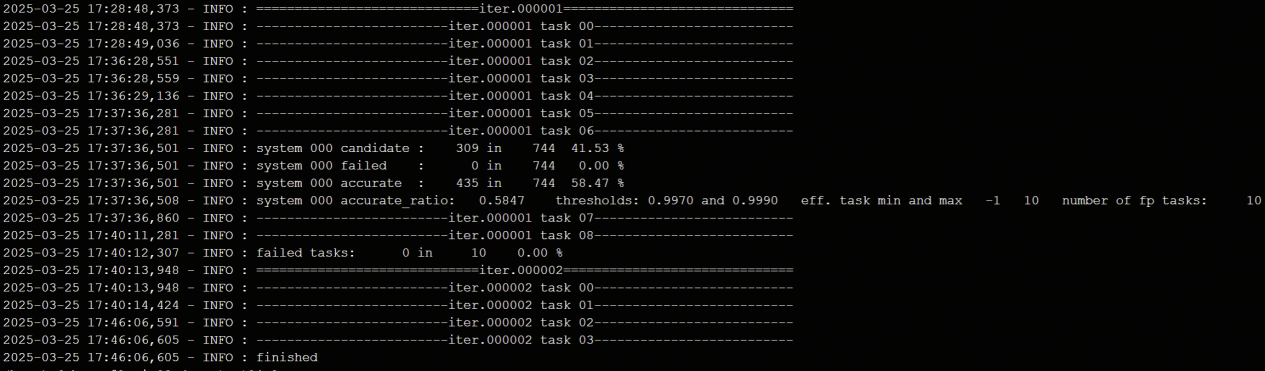
作业完成后,计算结果保存在当前文件夹中的./OUT.ABACUS文件夹中,查看running_scf.log文件。可知,经过18次迭代后,电荷密度收敛“charge density convergence is achieved”,密度误差达到7.03711049754e-08,最终总能量为-19447.849293 eV。 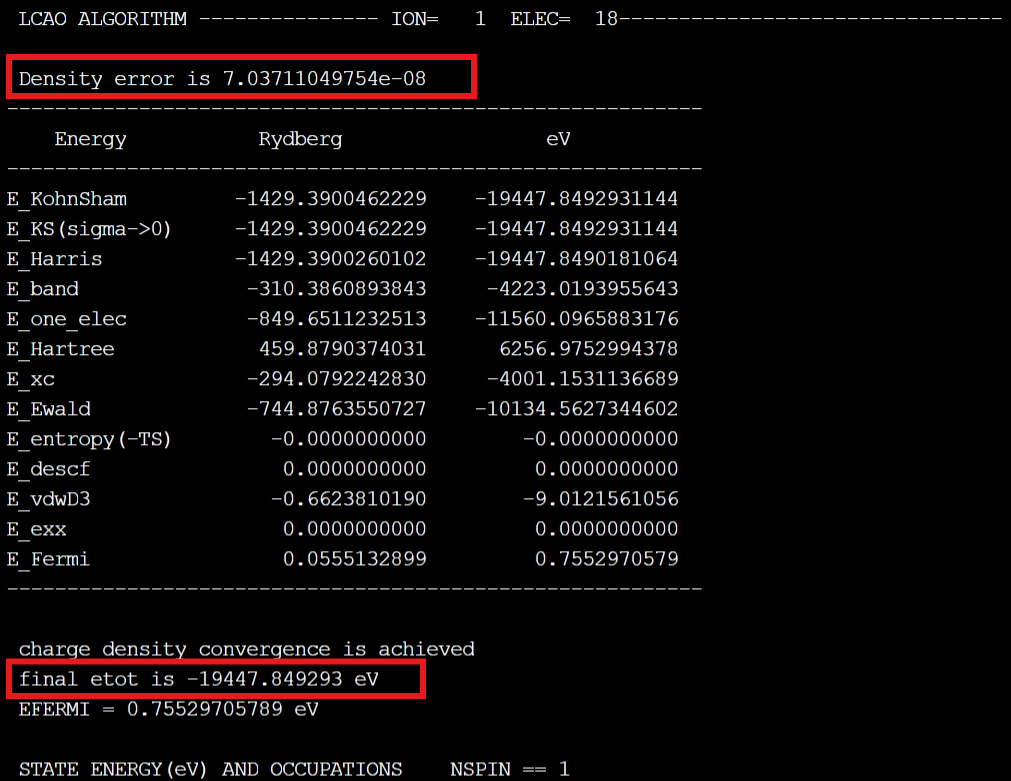
完成ABACUS第一性原理计算后,我们将使用Deepmd软件和ABACUS计算结果进行LiCl的深度势能模型训练。
cd进入deepmd-data文件夹,将我们提前准备好的文件上传,包括训练和验证数据、Deepmd训练脚本。使用“unzip 00.data”命令解压数据文件。
我们将ABACUS计算结果数据的80%作为训练数据(training_data),20%作为验证数据(validation_data)。00.data文件结构如下图: 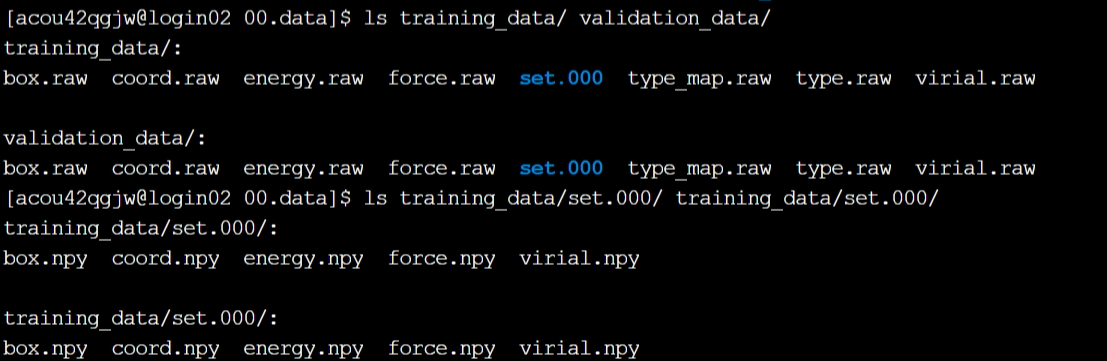 其中,set.000中包含压缩格式的数据(NumPy数组),文件以.npy为后缀;type.raw是原子类型文件;type_map.raw是原子类型名称文件;其它.raw为空间和物理信息文件。
其中,set.000中包含压缩格式的数据(NumPy数组),文件以.npy为后缀;type.raw是原子类型文件;type_map.raw是原子类型名称文件;其它.raw为空间和物理信息文件。
Deepmd模型训练需要提供输入脚本input.json,如下图: 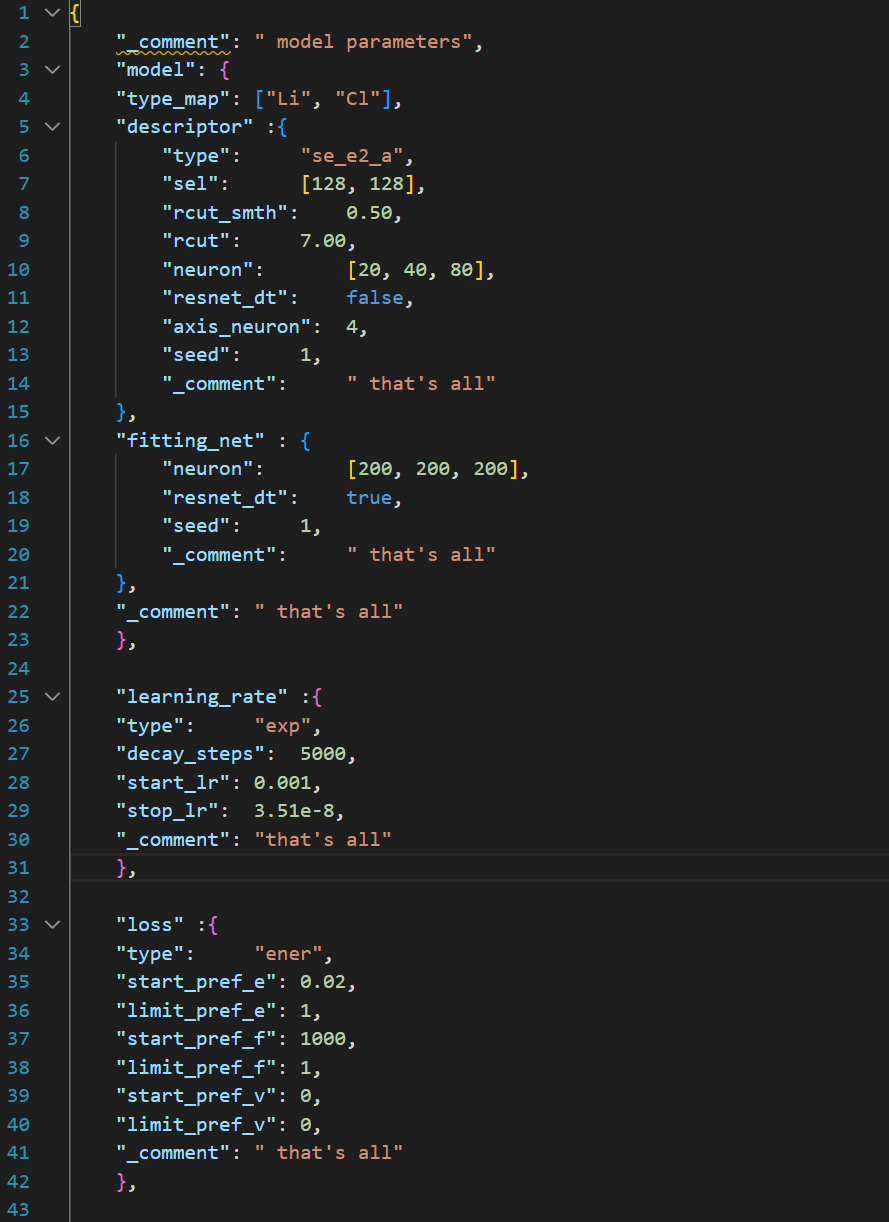 其中部分参数的含义如下表:
其中部分参数的含义如下表: 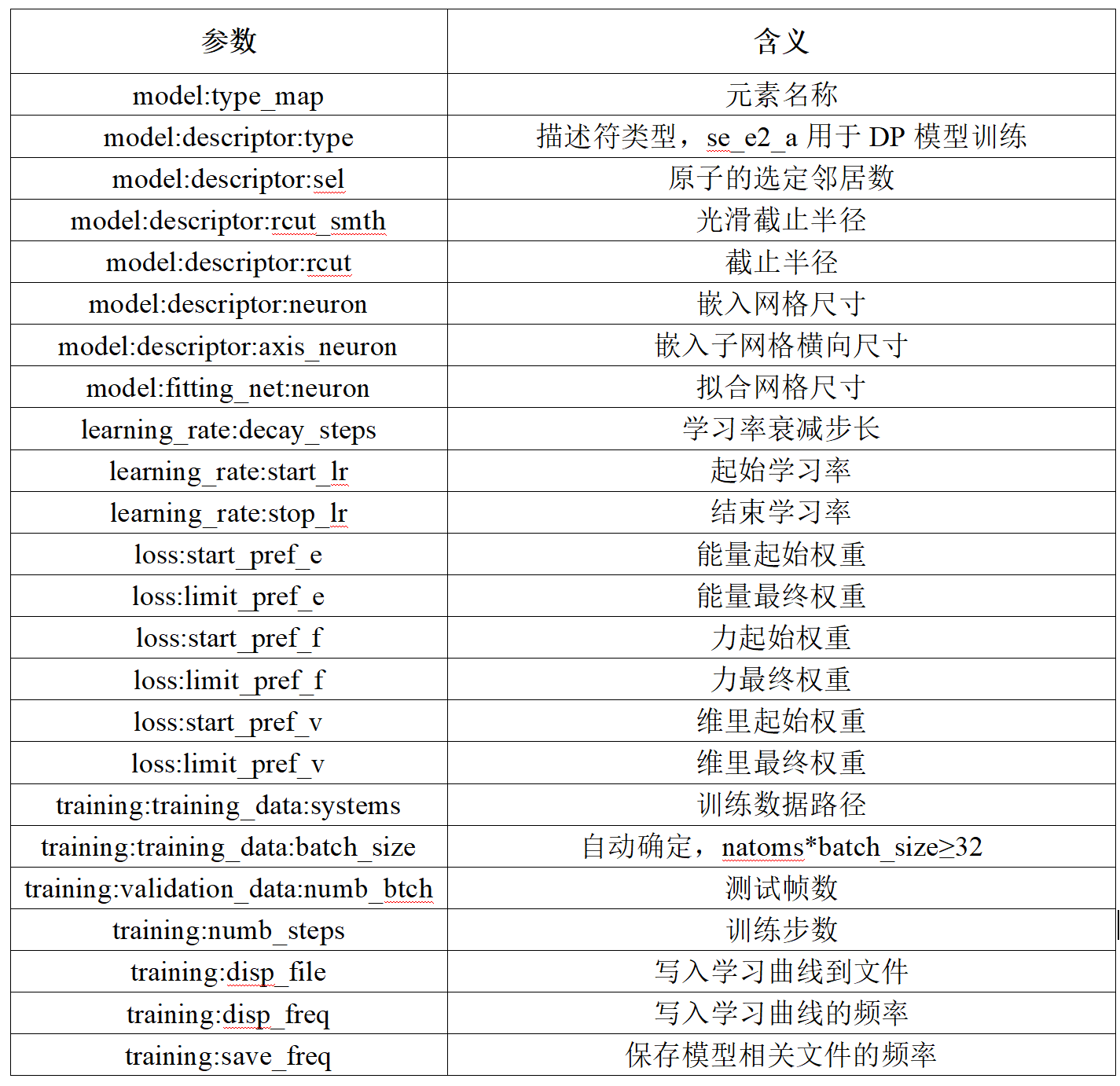 Deepmd软件申请成功后,默认安装在家目录的apprepo下,环境变量默认放在软件scripts目录下的env.sh中,通过修改case文件夹的脚本文件singluarity.slurm得到我们需要的脚本文件。我们使用单个节点的4个CPU核和4张异构加速卡AI进行计算,线程数为8。
Deepmd软件申请成功后,默认安装在家目录的apprepo下,环境变量默认放在软件scripts目录下的env.sh中,通过修改case文件夹的脚本文件singluarity.slurm得到我们需要的脚本文件。我们使用单个节点的4个CPU核和4张异构加速卡AI进行计算,线程数为8。
使用sbatch singularity.slurm提交计算任务;使用squeue查看作业情况;使用tail -f slurm-***.out动态查看软件运行输出。
每隔1万步,模型会保存在model.ckpt检查点文件中,训练和测试错误显示保存在lcurve.out文件中。
训练结束后,需要对模型进行冻结和压缩,压缩的DeepMD模型会降低内存消耗,加快计算速度。
修改singularity.slurm脚本,使用“dp freeze -o licl.pb”命令冻结模型,在当前路径下获得licl.pb。
使用“dp compress -i licl.pb -o licl-compress.pb”命令压缩模型,在当前路径下获得licl-compress.pb。
完成DeepMD深度势能模型训练之后,我们将使用已集成Lammps md计算的DeepMD软件基于训练的DP模型进行LiCl性质模拟,并利用经典分子动力学软件Lammps对模拟结果进行对比。
cd进入lammps-md-data文件夹,将我们提前准备好的文件上传,包括licl.data和licl.in。其中,licl.data是LiCl熔体MD模拟的初始配置,licl.in是lammps输入脚本。同时,需要将模型冻结生成的“licl-compress.pb”模型文件复制到当前路径下。文件情况如下图。 
申请的Deepmd v3.0.0b4加速版软件已集成lammps,默认安装在家目录的apprepo下,环境变量默认放在软件scripts目录下的env.sh中,通过修改case文件夹的脚本文件singluarity.slurm得到我们需要的脚本文件。我们使用单个节点的4个CPU核和4张异构加速卡AI进行计算,线程数为8。
使用sbatch singularity.slurm提交计算任务;使用squeue查看作业情况;使用tail -f slurm-***.out动态查看软件运行输出。
模拟结束后,在当前路径中生成licl.dump和log.lammps文件,分别存储热力学信息和分子轨迹。我们可以使用下面的Python脚本计算RDF并绘制图形。 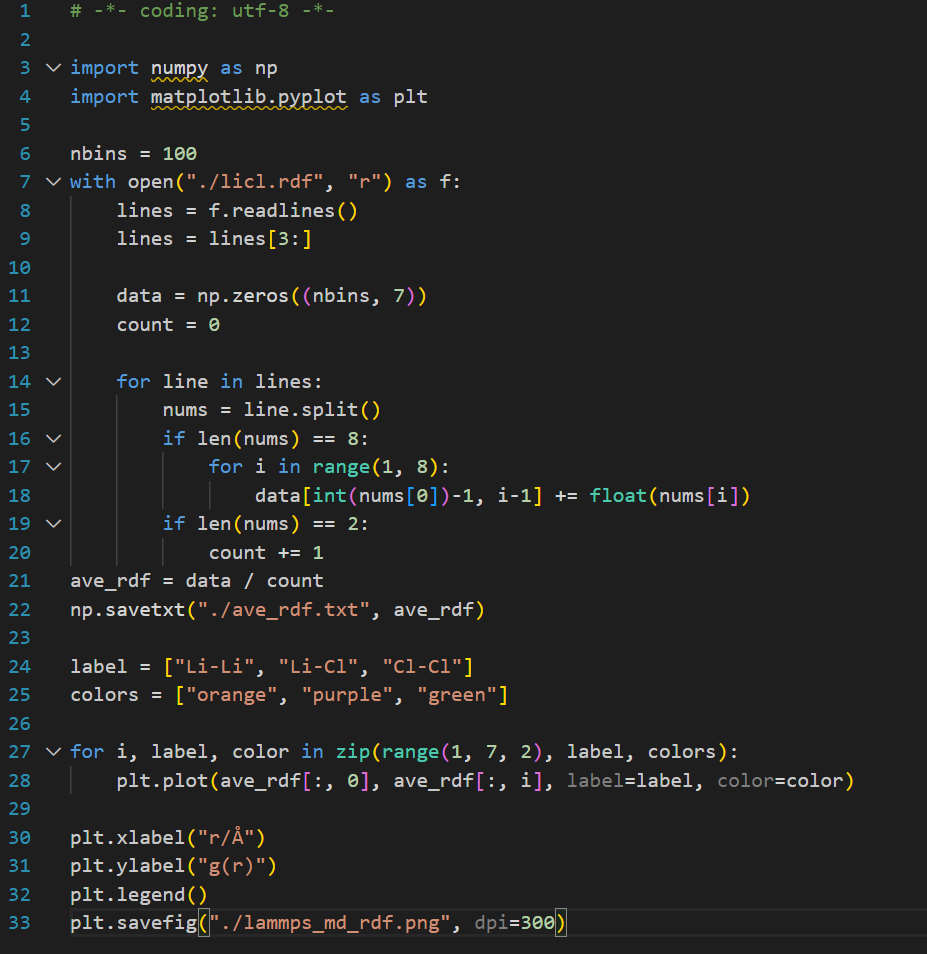 绘图步骤:
绘图步骤:
(1)使用“module load apps/anaconda3/5.2.0”命令加载anaconda3;
(2)使用“控制台-Linux桌面”申请图形化接口,如下图; 
 (3)使用“export DISPLAY=vncserver=01:1”配置环境变量;
(3)使用“export DISPLAY=vncserver=01:1”配置环境变量;
(4)使用“python rdf.py”绘图。 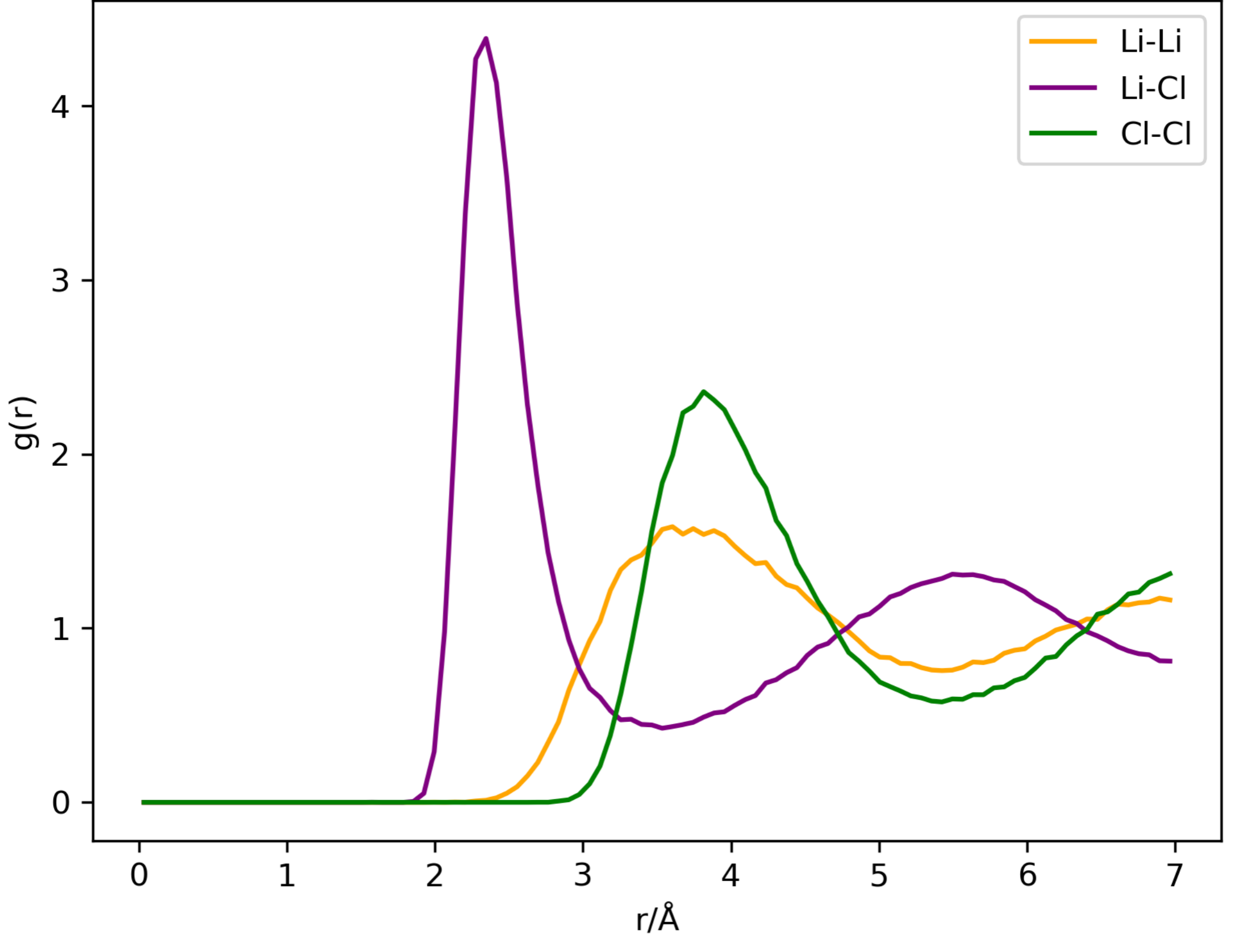 图:lammps-md模拟结果RDF曲线
图:lammps-md模拟结果RDF曲线
cd进入lammps-data文件夹,将我们提前准备好的文件上传,包括licl.data和licl.in。
软件申请成功后,默认安装在家目录的apprepo下,环境变量默认放在软件scripts目录下的env.sh中,通过修改case文件夹的脚本文件lammps.slurm得到我们需要的脚本文件。我们使用单个节点的32个CPU核。
使用sbatch lammps.slurm提交计算任务;使用squeue查看作业情况;使用tail -f slurm-***.out动态查看软件运行输出。
模拟结束后,在当前路径中生成log.lammps、licl.dump、licl.rdf、licl.msd文件,分别存储热力学信息、分子轨迹、径向分布函数、均方位移。我们可以使用之前介绍的Python脚本和相同流程来计算RDF并绘制图形。 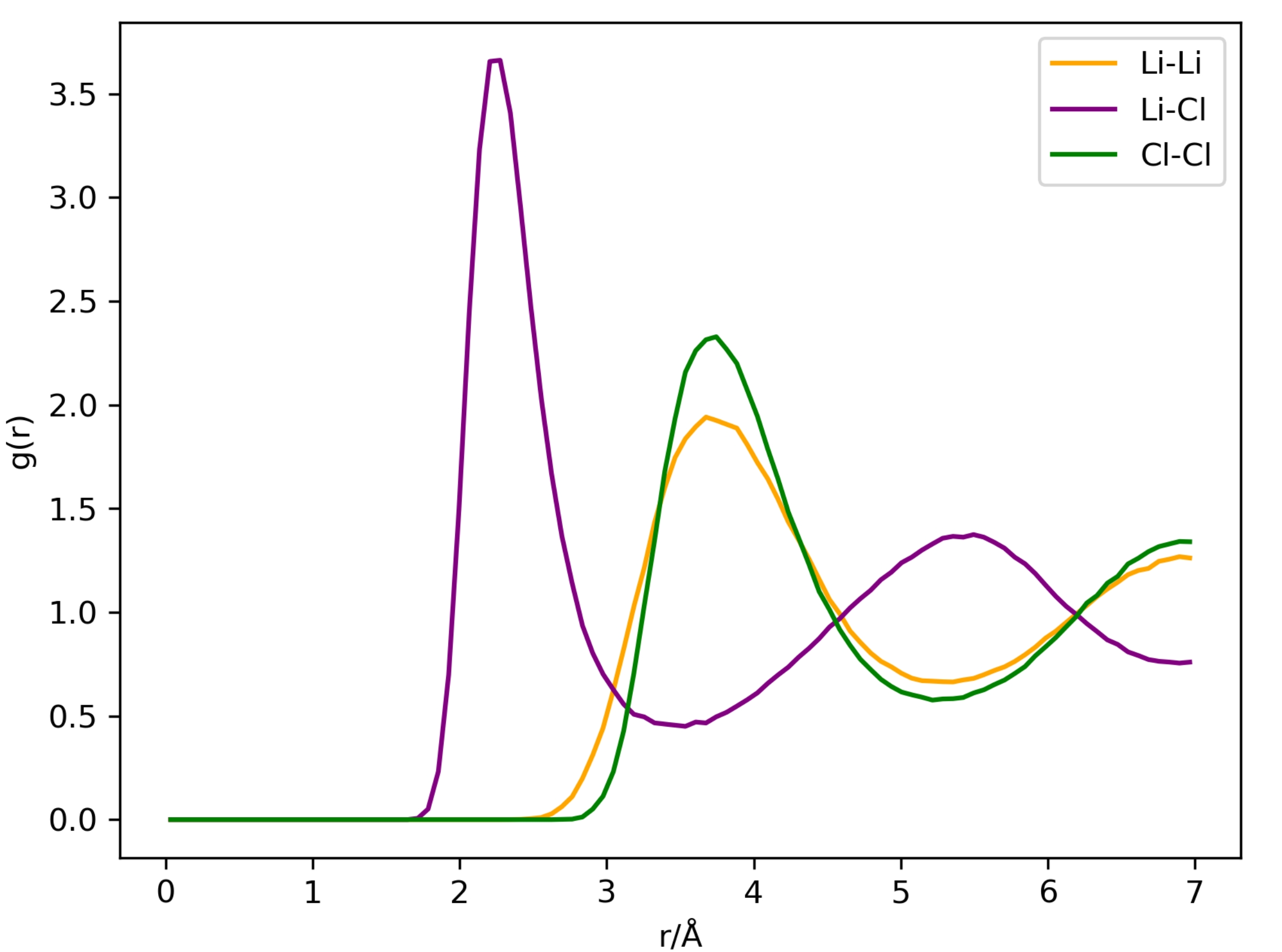 图:lammps模拟结果RDF曲线
图:lammps模拟结果RDF曲线
同时,我们可以使用下面的Python脚本msd.py利用均方位移(MSD)计算扩散系数: 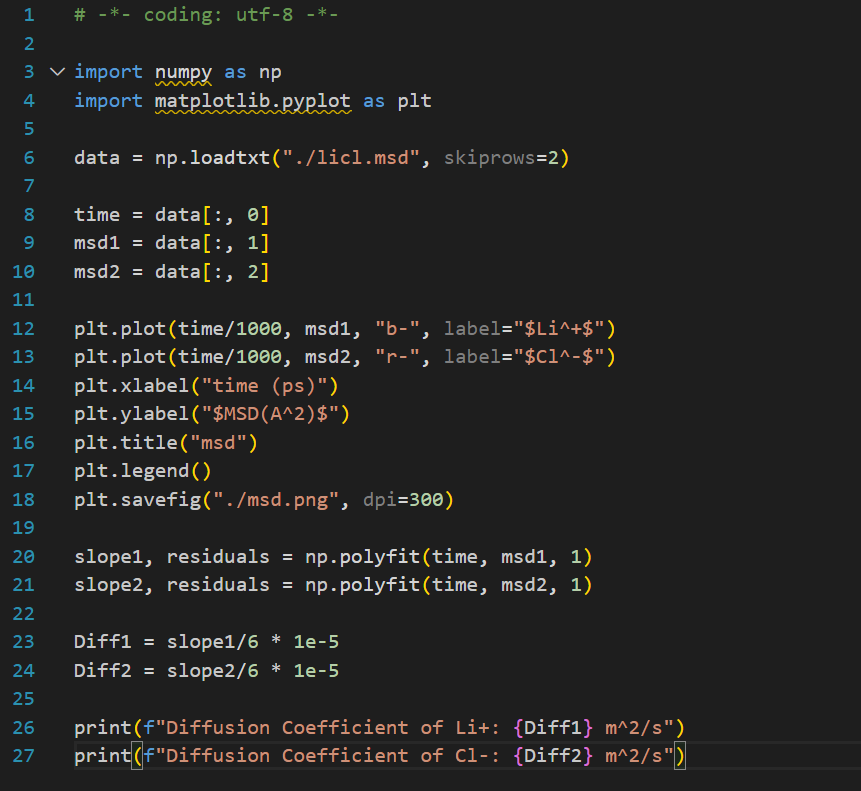
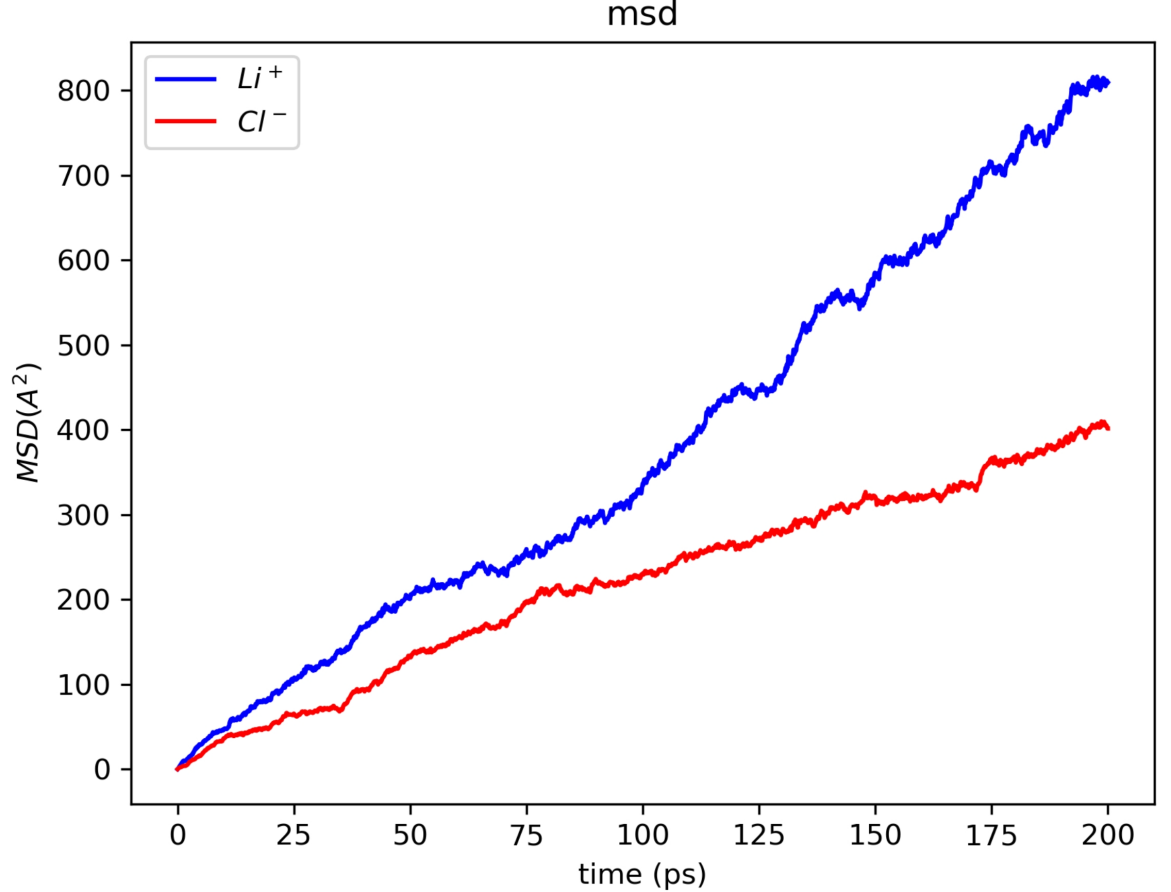 图:均方位移
图:均方位移
我们对基于第一性原理ABACUS+深度势能DeepMD训练的势函数的模拟结果与传统经典分子动力学软件Lammps的模拟结果进行对比。 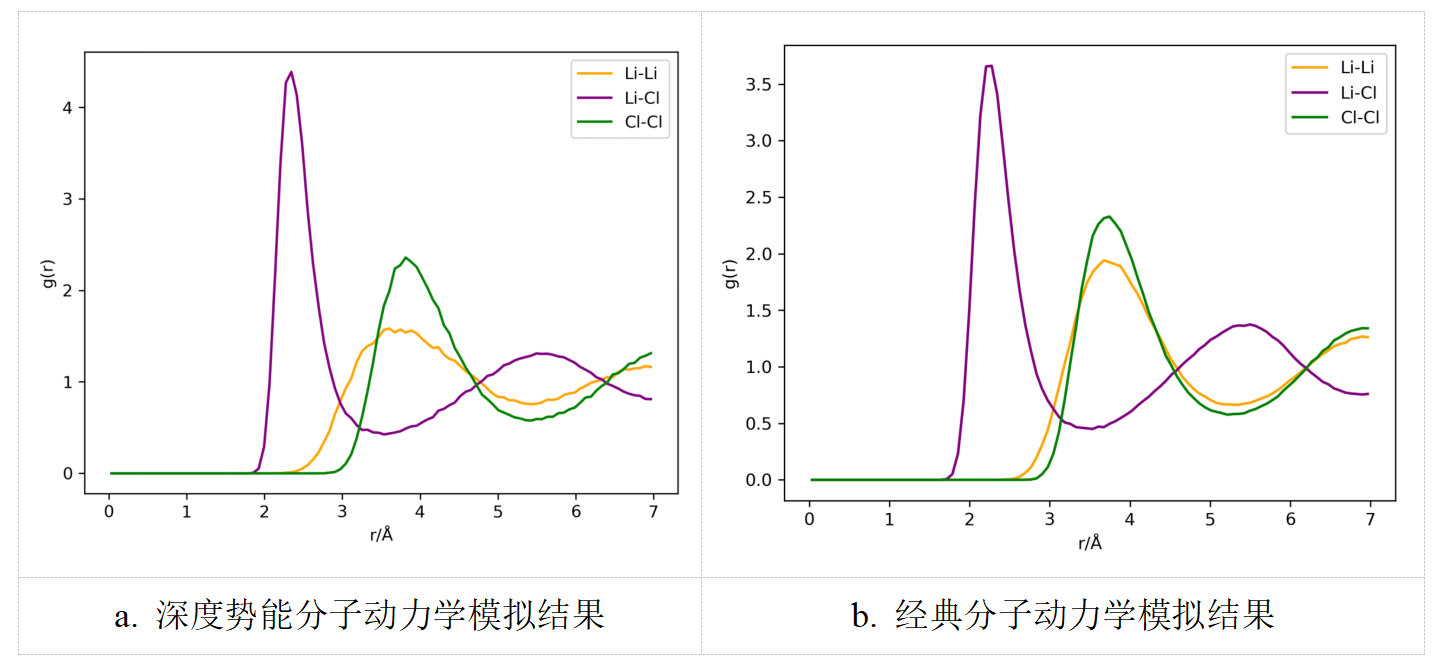 对比可知,两者的计算结果趋势一致,可以通过增加深度势能模型训练的训练步数、扩大训练数据集等方式来提升势函数精度。
对比可知,两者的计算结果趋势一致,可以通过增加深度势能模型训练的训练步数、扩大训练数据集等方式来提升势函数精度。
在超算互联网平台使用异构加速卡资源时,推荐ABACUS版本为:ABACUS LTS v3.10.0 异构加速版本

使用该异构版本推荐参考的INPUT参数设置文件:
C++
calculation scf
device gpu //选择计算设备
basis_type pw //仅支持PW基组
symmetry 0
ecutwfc 100
scf_thr 1e-08
cal_force 1
cal_stress 1
kspacing 0.08
pseudo_rcut 10
pseudo_mesh 1
ks_solver dav_subspace //推荐使用该对角化算法
pw_diag_ndim 2
pw_diag_nmax 20
pw_seed 0
relax_nmax 100
force_thr 0.001
stress_thr 0.5
smearing_method gaussian
smearing_sigma 0.01
mixing_beta 0.2
scf_nmax 120使用CPU资源时,推荐的ABACUS版本为:abacus-v3.10.0LTS

这是一个在超算互联网平台上使用dpgen + ABACUS进行采样的例子,算例包含以下文件:
运行计算:
# 激活dpgen conda环境,如果你的环境已经安装好了dpgen和dpdata,不需要执行这步
source ~/miniconda3/bin/activate dpgen-dpdata
# 运行dpgen
dpgen run run_param.json machine.json &运行结束后,会在dpgen.log文件中,出现类似如下的输出: