高性能计算服务
>
软件专区
>
最佳实践
>
利用CP2K解析Li10GeP2S12固态电解质中锂离子迁移机制
CP2K软件中的从头算分子动力学(AIMD)方法,可实时计算原子受力,直接模拟体系动态演化过程。
相比传统静态计算(能垒搜索+电子结构分析),AIMD模拟可实时捕获化学键断裂/形成过程,揭示离子迁移的协同机制与瞬态中间态结构,为材料动力学行为提供直接证据。且无需预设势函数参数,避免ReaxFF等反应力场的经验性约束,显著提升复杂体系(如界面反应、相变过程)的模拟普适性。
本次实操,基于超算互联网平台,通过CP2K软件对固态电解质Li10GeP2S12 (LGPS) 的锂离子迁移行为进行AIMD模拟,计算锂离子扩散系数及迁移势垒。
超算互联网提供多种编译好的CP2K,支持开箱即用。本次实操,我们使用cp2k-v2025.1-oneapi2024版本,如下图所示: 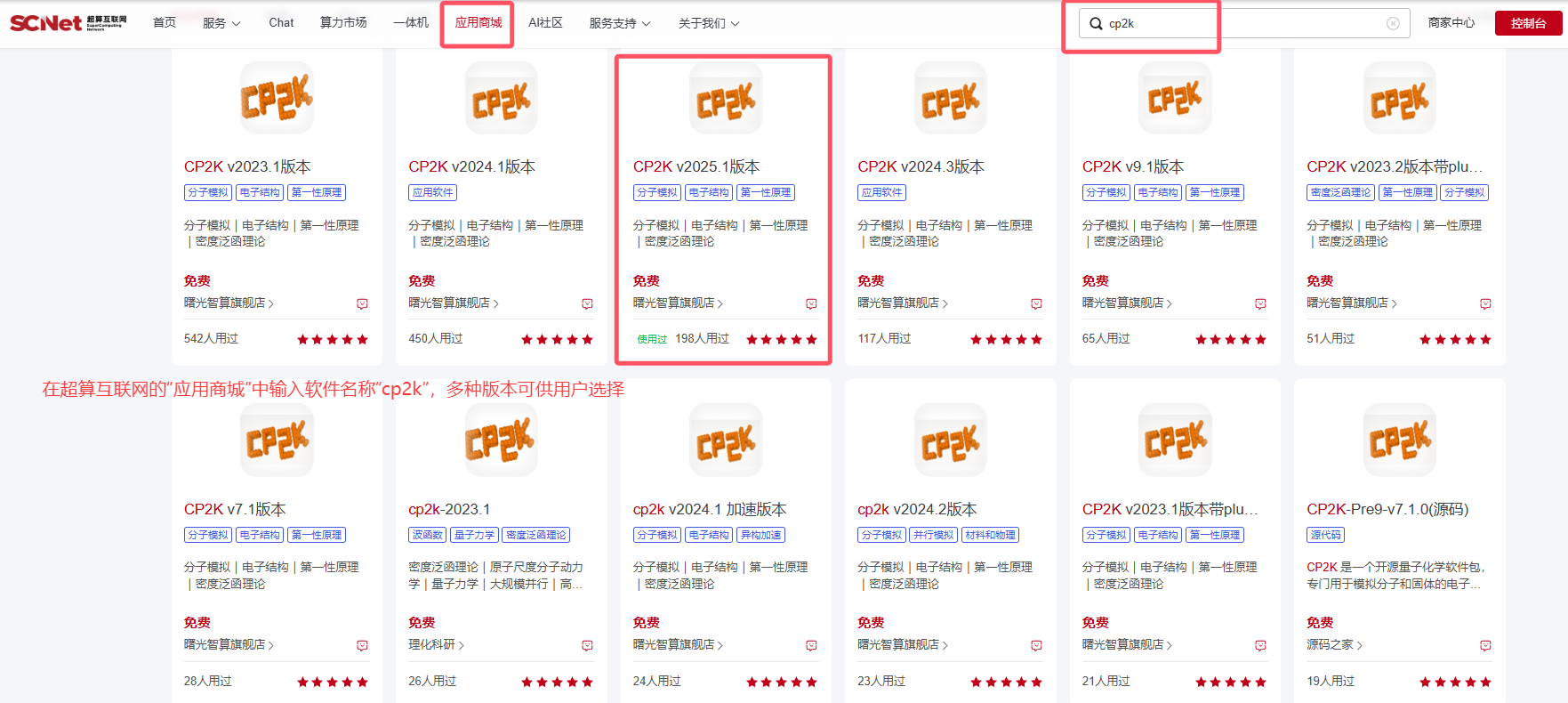 结构优化是DFT计算必不可少的第一步,稳定的构型对后续的模拟至关重要。首先将结构文件POSCAR.cif上传至文件夹opt中,用超算互联网提供的Multiwfn软件准备cp2k.inp文件对其进行结构优化。
结构优化是DFT计算必不可少的第一步,稳定的构型对后续的模拟至关重要。首先将结构文件POSCAR.cif上传至文件夹opt中,用超算互联网提供的Multiwfn软件准备cp2k.inp文件对其进行结构优化。 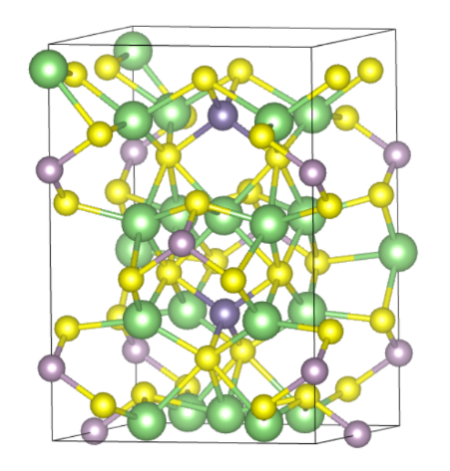
 按照提示输入目录:/public/home/lml1234/LGPS/opt/POSCAR.cif。调用Multiwfn程序。
按照提示输入目录:/public/home/lml1234/LGPS/opt/POSCAR.cif。调用Multiwfn程序。 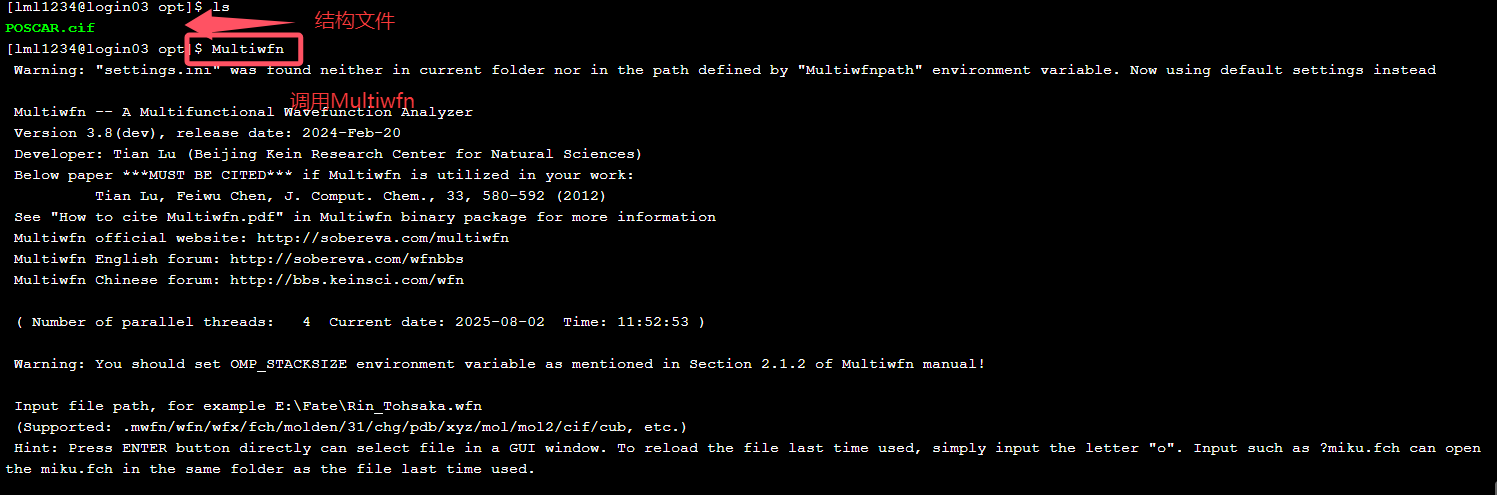 依次输入:cp2k-enter ——-1-3-6-8-3,3,2,就会在当前文件夹生成结构优化的输入文件POSCAR.inp
依次输入:cp2k-enter ——-1-3-6-8-3,3,2,就会在当前文件夹生成结构优化的输入文件POSCAR.inp
#Generated by Multiwfn
@SET DATAPATH /public/home/lml1234/apprepo/cp2k/v2025.1-oneapi2024/app/data
&GLOBAL
PROJECT POSCAR
PRINT_LEVEL LOW
RUN_TYPE GEO_OPT
&END GLOBAL
&FORCE_EVAL
METHOD Quickstep
&SUBSYS
&CELL
A 8.78765000 0.00000000 0.00000000
B 0.00000000 8.78765000 0.00000000
C 0.00000000 0.00000000 12.65755000
PERIODIC XYZ #Direction(s) of applied PBC (geometry aspect)
&END CELL
&COORD
Li 2.04007931 2.38843933 3.75933032
Li 6.74750039 6.39893825 3.75962145
Li 6.39892068 2.04021113 10.08837113
Li 2.38842176 6.74764099 10.08813064
Li 2.04021113 6.39892068 3.75959613
Li 6.74764099 2.38842176 3.75935564
Li 6.39893825 6.74750039 10.08839645
Li 2.38843933 2.04007052 10.08810532
Li 2.25261741 2.43548841 0.50509953
Li 6.53505016 6.35213523 0.50518814
Li 6.35211765 2.25263499 6.83392516
Li 2.43547083 6.53506774 6.83392516
Li 2.25263499 6.35211765 0.50515016
Li 6.53506774 2.43547083 0.50515016
Li 6.35213523 6.53505016 6.83396314
Li 2.43548841 2.25261741 6.83387453
Li 0.00021969 8.78763242 5.53522256
Li 8.78763242 0.00021969 11.86399756
Li 4.39383379 4.39379864 0.61854915
Li 4.39379864 4.39383379 6.94732415
Ge 4.39384258 4.39381621 3.81556782
Ge 4.39381621 4.39384258 10.14434282
P 8.78764121 8.78756212 2.31904037
P 8.78756212 8.78764121 8.64781537
P 0.00005273 4.39378985 6.34977388
P 4.39378985 0.00005273 0.02099888
S 8.78757970 1.68675427 9.80576601
S 8.78763242 7.10085179 9.80579133
S 7.10086058 8.78763242 3.47701633
S 1.68675427 8.78757970 3.47699101
S 0.00001758 1.71225603 1.15197628
S 0.00001758 7.07565760 1.15130543
S 7.07565760 0.00001758 7.48008043
S 1.71225603 0.00001758 7.48075128
S 0.00007909 2.68570796 5.18939298
S 0.00007909 6.10190689 5.18936766
S 6.10190689 0.00007909 11.51814266
S 2.68570796 0.00007909 11.51816798
S 1.66801900 4.39382500 7.53531798
S 7.11964858 4.39382500 7.53515343
S 4.39382500 1.66801900 1.20654298
S 4.39382500 7.11964858 1.20637843
S 2.54387528 4.39383379 11.44195687
S 6.24377472 4.39383379 11.44195687
S 4.39383379 2.54387528 5.11318187
S 4.39383379 6.24377472 5.11318187
S 2.56304994 4.39381621 2.51337173
S 6.22463521 4.39381621 2.51339705
S 4.39381621 2.56304994 8.84214673
S 4.39381621 6.22463521 8.84217205
&END COORD
&KIND Li
ELEMENT Li
BASIS_SET DZVP-MOLOPT-SR-GTH-q3
POTENTIAL GTH-PBE
&END KIND
&KIND Ge
ELEMENT Ge
BASIS_SET DZVP-MOLOPT-SR-GTH-q4
POTENTIAL GTH-PBE
&END KIND
&KIND P
ELEMENT P
BASIS_SET DZVP-MOLOPT-SR-GTH-q5
POTENTIAL GTH-PBE
&END KIND
&KIND S
ELEMENT S
BASIS_SET DZVP-MOLOPT-SR-GTH-q6
POTENTIAL GTH-PBE
&END KIND
&END SUBSYS
&DFT
BASIS_SET_FILE_NAME ${DATAPATH}/BASIS_MOLOPT
POTENTIAL_FILE_NAME ${DATAPATH}/POTENTIAL
CHARGE 0 #Net charge
MULTIPLICITY 1 #Spin multiplicity
&KPOINTS
SCHEME MONKHORST-PACK 3 3 2
&END KPOINTS
&QS
EPS_DEFAULT 1.0E-12 #Set all EPS_xxx to values such that the energy will be correct up to this value
# EXTRAPOLATION USE_PREV_P #Use converged density matrix of last geometry as initial guess
&END QS
&POISSON
PERIODIC XYZ #Direction(s) of PBC for calculating electrostatics
PSOLVER PERIODIC #The way to solve Poisson equation
&END POISSON
&XC
&XC_FUNCTIONAL PBE
&END XC_FUNCTIONAL
&END XC
&MGRID
CUTOFF 400
REL_CUTOFF 55
&END MGRID
&SCF
MAX_SCF 128
EPS_SCF 1.0E-06 #Convergence threshold of density matrix of inner SCF
# SCF_GUESS RESTART #Use wavefunction from WFN_RESTART_FILE_NAME file as initial guess
# IGNORE_CONVERGENCE_FAILURE #Continue calculation even if SCF not converged, works for version >= 2024.1
&DIAGONALIZATION
ALGORITHM STANDARD #Algorithm for diagonalization
&END DIAGONALIZATION
&MIXING #How to mix old and new density matrices
METHOD BROYDEN_MIXING #PULAY_MIXING is also a good alternative
ALPHA 0.4 #Default. Mixing 40% of new density matrix with the old one
NBROYDEN 8 #Default is 4. Number of previous steps stored for the actual mixing scheme
&END MIXING
&SMEAR
METHOD FERMI_DIRAC
ELECTRONIC_TEMPERATURE 300 #Electronic temperature of Fermi-Dirac smearing in K
&END SMEAR
ADDED_MOS 30 #Number of virtual MOs to solve
&PRINT
&RESTART #Note: Use "&RESTART OFF" can prevent generating .wfn file
BACKUP_COPIES 0 #Maximum number of backup copies of wfn file. 0 means never
&END RESTART
&END PRINT
&END SCF
&END DFT
&END FORCE_EVAL
&MOTION
&GEO_OPT
TYPE MINIMIZATION #Search for minimum
KEEP_SPACE_GROUP F #If T, then space group will be detected and preserved
OPTIMIZER BFGS #Can also be CG (more robust for difficult cases) or LBFGS
&BFGS
TRUST_RADIUS 0.2 #Trust radius (maximum stepsize) in Angstrom
# RESTART_HESSIAN T #If read initial Hessian, uncomment this line and specify the file in the next line
# RESTART_FILE_NAME to_be_specified
&END BFGS
MAX_ITER 500 #Maximum number of geometry optimization
MAX_DR 3E-3 #Maximum geometry change
RMS_DR 1.5E-3 #RMS geometry change
MAX_FORCE 4.5E-4 #Maximum force
RMS_FORCE 3E-4 #RMS force
&END GEO_OPT
&PRINT
&TRAJECTORY
FORMAT xyz
&END TRAJECTORY
&RESTART
BACKUP_COPIES 0 #Maximum number of backing up restart file, 0 means never
&END RESTART
&END PRINT
&END MOTION小技巧:常规操作中,我们需要将基组文件BASIS_MOLOPT和赝势文件POTENTIALS拷贝到我们计算文件夹中,在本例中,利用linux中的set命令使用绝对路径指定将其写入输入文件中,计算的时候直接引用即可。
添加一行文件所在路径:@SET DATAPATH /public/home/lml1234/apprepo/cp2k/v2025.1-oneapi2024/app/data 
(1) 输入文件:采用结构优化得到的稳定构型进行AIMD模拟,这里仍旧命名为POSCAR.cif,准备输入文件cp2k.inp文件。
@SET DATAPATH /public/home/lml1234/apprepo/cp2k/v2025.1-oneapi2024/app/data
&GLOBAL
PROJECT cp2k
RUN_TYPE MD
PRINT_LEVEL LOW
&END GLOBAL
&FORCE_EVAL
METHOD Quickstep
&DFT
BASIS_SET_FILE_NAME ${DATAPATH}/BASIS_MOLOPT
POTENTIAL_FILE_NAME ${DATAPATH}/GTH_POTENTIALS
WFN_RESTART_FILE_NAME ./cp2k-RESTART.wfn
UKS T #LSD
&MGRID
NGRIDS 4
CUTOFF 400
REL_CUTOFF 60
&END MGRID
&QS
WF_INTERPOLATION ASPC
EXTRAPOLATION_ORDER 3
&END QS
&XC
&XC_FUNCTIONAL PBE
&END XC_FUNCTIONAL
&END XC
&SCF
SCF_GUESS RESTART
MAX_SCF 100
EPS_SCF 1.0E-06
&OT T
MINIMIZER CG
LINESEARCH 2PNT
PRECONDITIONER FULL_ALL
&END OT
&OUTER_SCF T
EPS_SCF 1.0E-05
MAX_SCF 100
&END OUTER_SCF
&END SCF
&END DFT
&SUBSYS
&CELL
ABC 8.78765 8.78765 12.65755
ALPHA_BETA_GAMMA 90 90 90
&END CELL
&TOPOLOGY
COORD_FILE_NAME POSCAR.cif
COORD_FILE_FORMAT cif
&END TOPOLOGY
&KIND Ge
BASIS_SET DZVP-MOLOPT-SR-GTH
POTENTIAL GTH-PBE-q4
&END KIND
&KIND P
BASIS_SET DZVP-MOLOPT-SR-GTH
POTENTIAL GTH-PBE-q5
&END KIND
&KIND S
BASIS_SET DZVP-MOLOPT-SR-GTH
POTENTIAL GTH-PBE-q6
&END KIND
&KIND Li
BASIS_SET DZVP-MOLOPT-SR-GTH
POTENTIAL GTH-PBE-q3
&END KIND
&END SUBSYS
&END FORCE_EVAL
&MOTION
&MD
ENSEMBLE NVT
STEPS 20000
TIMESTEP 1
TEMPERATURE 600.0
&THERMOSTAT
TYPE NOSE
&NOSE
LENGTH 3
YOSHIDA 3
MTS 2
TIMECON [wavenumber_t] 1000
&END NOSE
&END THERMOSTAT
&END MD
&PRINT
&TRAJECTORY
&EACH
MD 1
&END EACH
&END TRAJECTORY
&VELOCITIES
&EACH
MD 1
&END EACH
&END VELOCITIES
&RESTART_HISTORY
&EACH
MD 500
&END EACH
&END RESTART_HISTORY
&END PRINT
&END MOTION关于输入文件cp2k.inp的几点说明:
GLOBAL部分: RUN_TYPE为MD,同时设置PRINT_LEVEL为LOW减少输出从而减轻服务器的负担。  MOTION部分:设置系综、温度、步长(默认单位fs)、步数,选择NVT系综,并设置Nose-Hoover恒温器来维持恒定的温度。因为AIMD模拟可随时手动停止,所以步长可以设置长一点,通过观察输出的轨迹满足期待的效果时即可停止。
MOTION部分:设置系综、温度、步长(默认单位fs)、步数,选择NVT系综,并设置Nose-Hoover恒温器来维持恒定的温度。因为AIMD模拟可随时手动停止,所以步长可以设置长一点,通过观察输出的轨迹满足期待的效果时即可停止。  (2)结果分析: -pos-1.xyz文件中记录了原子的位置信息,这里利用超算互联网中提供的vmd软件对锂离子迁移路径进行可视化处理,将输出文件导入到vmd软件。
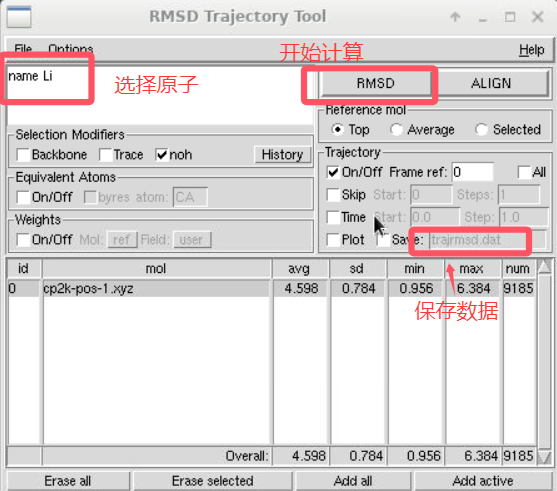
(2)结果分析: -pos-1.xyz文件中记录了原子的位置信息,这里利用超算互联网中提供的vmd软件对锂离子迁移路径进行可视化处理,将输出文件导入到vmd软件。 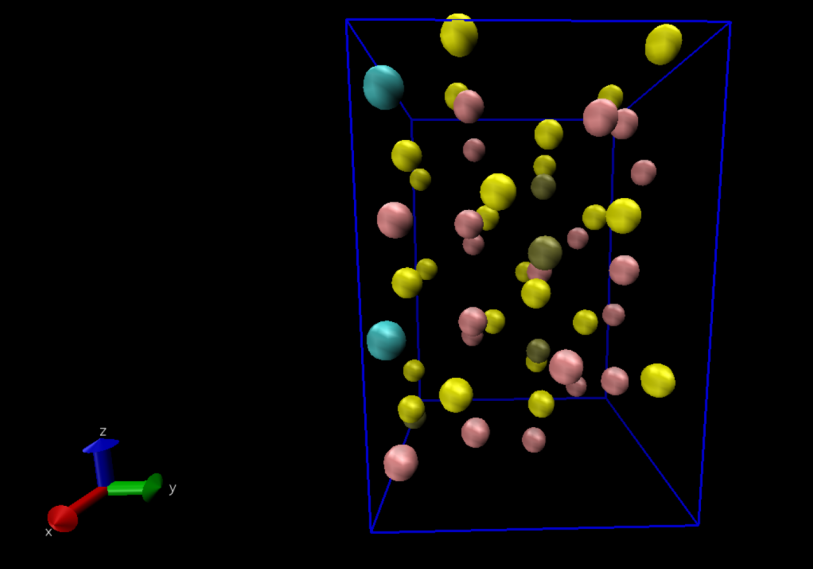 进行如下图所示的操作后即可得到锂离子的轨迹迁移路径。
进行如下图所示的操作后即可得到锂离子的轨迹迁移路径。 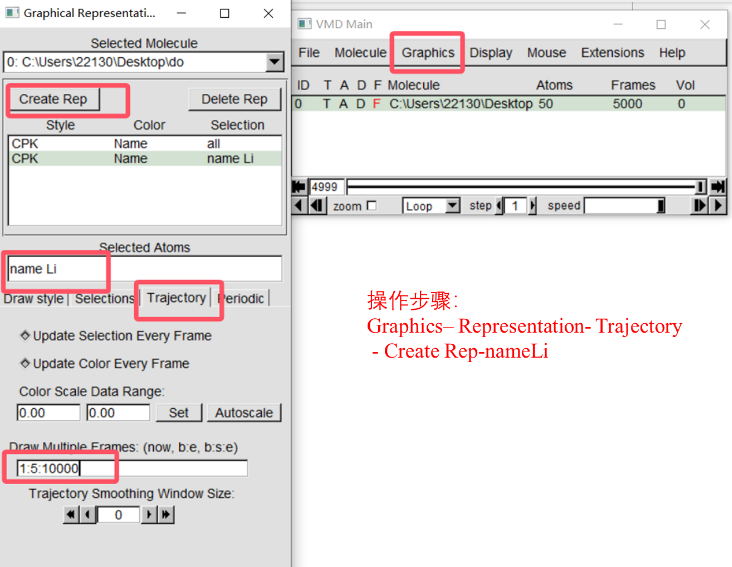
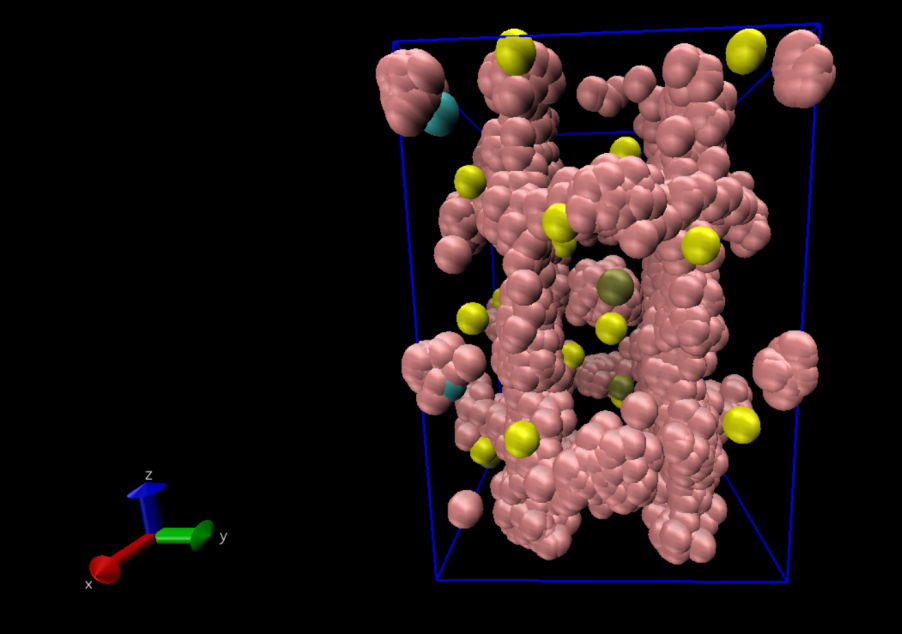 通过对模拟过程中Li的迁移轨迹可视化处理我们能够得出结论,Li离子沿C轴方向进行了迁移。接下来进行数据分析。
通过对模拟过程中Li的迁移轨迹可视化处理我们能够得出结论,Li离子沿C轴方向进行了迁移。接下来进行数据分析。
均方位移(Mean square displacement, MSD )是相对于参考位置的粒子位置随时间变化的度量。VMD可以计算方均根位移(Root Mean square displacement),对其进行平方即可得到MSD。 
 扩散常数(diffusion coefficient)是MD模拟描述离子/分子迁移性质的重要参数,通过AIMD求算扩散常数常用的是Einstein公式:
扩散常数(diffusion coefficient)是MD模拟描述离子/分子迁移性质的重要参数,通过AIMD求算扩散常数常用的是Einstein公式: 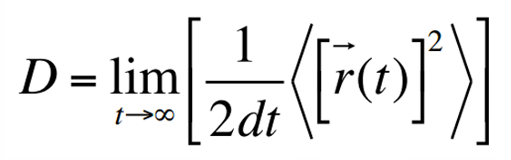 通过该公式求得在600K时,Li离子的扩散常数为7.41×10^-6。
通过该公式求得在600K时,Li离子的扩散常数为7.41×10^-6。
扩散系数和能垒关系满足,阿伦尼乌斯公式: 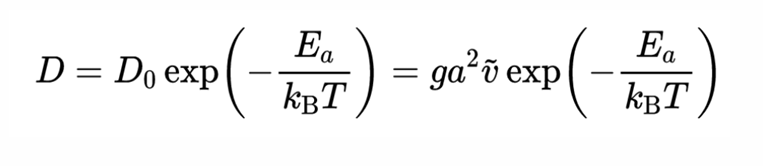 最终求得Li离子的迁移能垒为Ea=0.214 eV。
最终求得Li离子的迁移能垒为Ea=0.214 eV。