高性能计算服务
>
软件专区
>
最佳实践
>
基于Pymatgen的态密度分析全流程实战
在材料模拟研究中,态密度计算是关联电子结构与宏观物性的核心环节,而传统手动操作流程存在效率低、易出错、数据可比性差等痛点。
Pymatgen(Python Materials Genomics)是一个功能强大的开源 Python 库,用于材料分析。
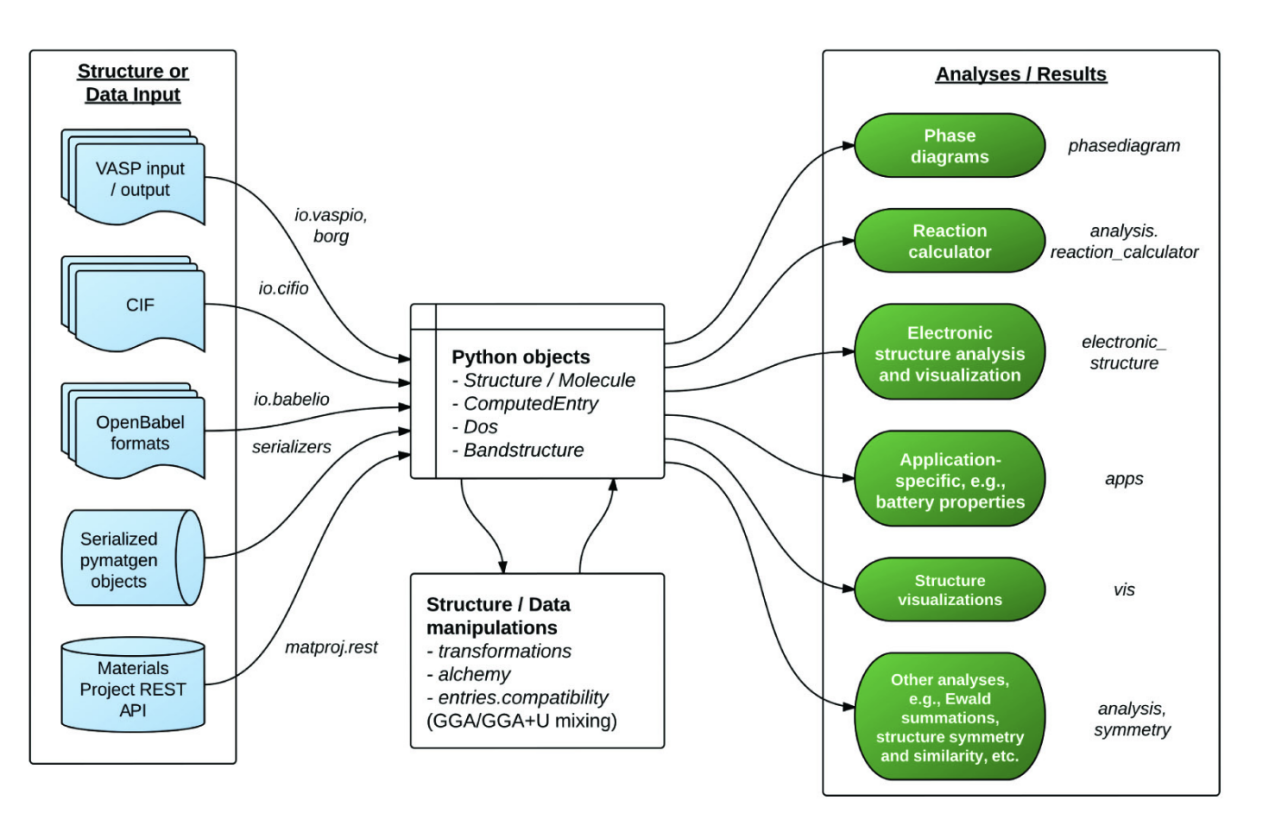
其主要功能特点包括:
本次实操,我们将在超算互联网使用Pymatgen实现从输入文件生成、态密度后处理,到可视化绘图的全流程,为催化材料设计、半导体能带态密度优化等场景提供高效解决方案。
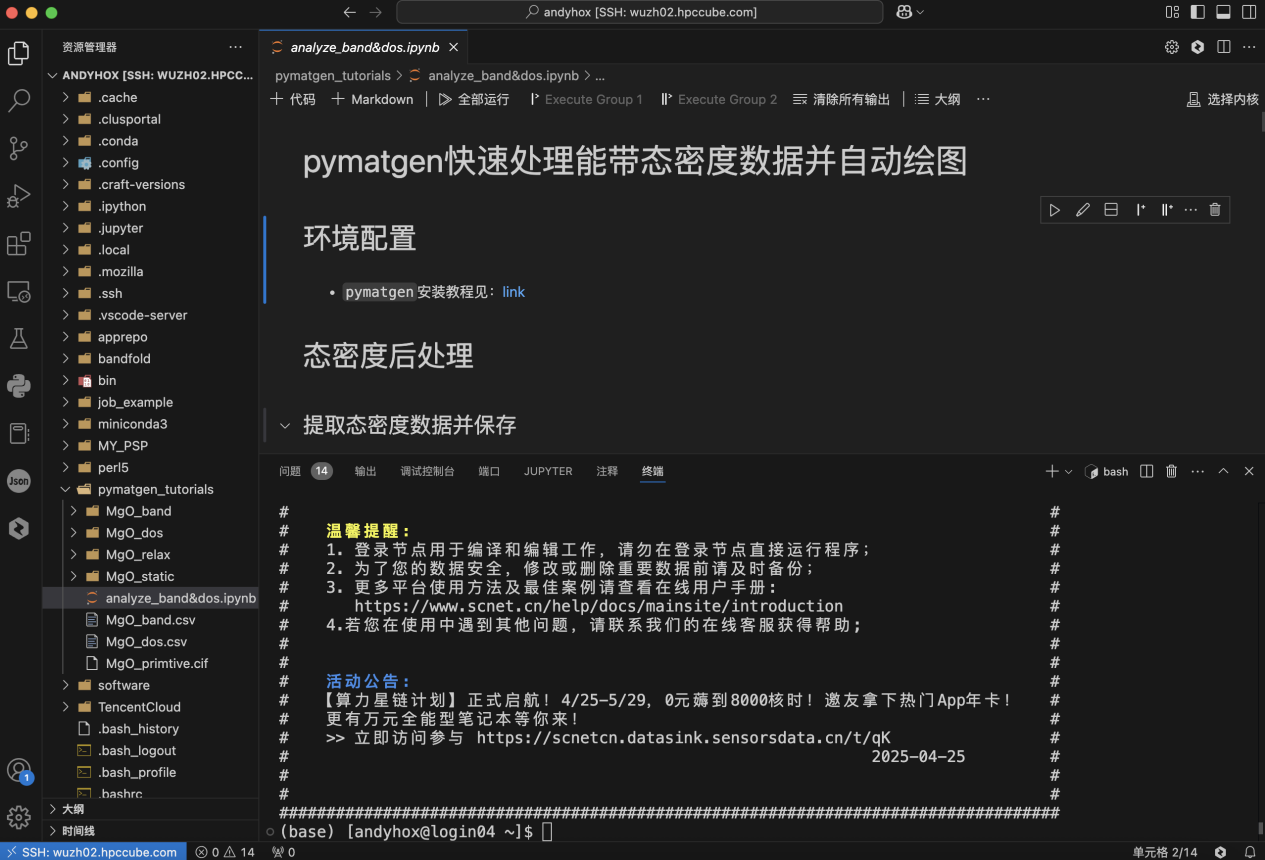
Pymatgen官网提供了详细的安装步骤,在超算互联网可以采用密钥的方式通过VSCode连接到对应的集群,官网给出了详细的配置教程: https://www.scnet.cn/help/docs/mainsite/hpc/cmd/connect-to-hpc/
通过VSCode连接集群进行操作的优点:
边写代码,边跑命令——分屏操作美滋滋
告别vim地狱,图形化编辑超直观
实时同步文件,再也不用本地→集群来回倒
完全替代Xshell+WinSCP组合!
晶体结构态密度的计算需要经过三个步骤:
常规计算操作基于vim进行文件编译,针对不同的计算修改INCAR参数;也有其他非常好用的工具,如vaspkit可以根据不同的计算类别自动生成计算的输入文件。
本次实操,我们介绍另一种自动生成计算输入文件的办法——基于Pymatgen自动生成计算文件,且可以自定义计算参数。
这里主要介绍三个类方法:MPRelaxSet、MPStaticSet、MPNonSCFSet,分别对应自动生成结构优化、静态自洽以及非自洽计算的输入文件。
MPRelaxSet有默认的计算参数,因此不需要设置所有的参数,只需要根据不同的体系设置对应的参数即可,代码为:
# 导入包
from pymatgen.core import Structure
from pymatgen.io.vasp.sets import MPRelaxSet
import os
# 工作目录
work_dir = '/work/home/andyhox/pymatgen_tutorials'
# 读取结构
struct = Structure.from_file(os.path.join(work_dir,'MgO_primtive.cif'))
# 设置输入set
relax_set = MPRelaxSet(
structure=struct, # 读取结构
user_incar_settings={"ENCUT":520, "ALGO":"Normal",
"EDIFFG":-0.01, "EDIFF":1e-6, "ISPIN":1}, #自定义INCAR参数
user_potcar_functional='PBE_54' # 定义泛函种类
)
# 保存输入文件
relax_set.write_input(os.path.join(work_dir,'MgO_relax'))运行代码后,会在当前的工作目录下生成一个MgO_relax文件夹,里面包含了自动生成的INCAR、KPOINTS、POSCAR、POTCAR四个文件。
NOTE:在此之前一定要配置好pymatgen,特别是配置好势函数,教程见官网。
在结构优化的基础上,可以用MPStaticSet读取优化的结果生成新的输入文件:
# 导入包
from pymatgen.io.vasp.sets import MPStaticSet
import os
# 工作目录
work_dir = os.getcwd()
# 设置输入set
static_set = MPStaticSet.from_prev_calc(
prev_dir=os.path.join(work_dir, 'MgO_relax'), # 读取结构优化的文件夹
user_incar_settings={"EDIFF":1e-7}, #自定义INCAR参数
user_potcar_functional='PBE_54' # 定义泛函种类
)
# 保存输入文件
static_set.write_input(os.path.join(work_dir,'MgO_static'))MPStaticSet.from_prev_calc()方法可以直接读取优化后的结构和INCAR输入,并在此基础上生成自洽计算的INCAR输入。
基于静态自洽的结果,同样的采用MPNonSCFSet.from_prev_calc()读取静态自洽的计算结果,生成态密度计算输入文件:
# 导入包
from pymatgen.io.vasp.sets import MPNonSCFSet
import os
# 工作目录
work_dir = os.getcwd()
# 设置输入set
nonscf_set = MPNonSCFSet.from_prev_calc(
prev_calc_dir=os.path.join(work_dir, 'MgO_static'), # 读取结构优化的文件夹
mode='uniform', # 定义K点生成格式
user_potcar_functional='PBE_54' # 定义泛函种类
)
# 保存输入文件
nonscf_set.write_input(os.path.join(work_dir,'MgO_dos'))运行代码后在MgO_dos文件夹生成INCAR、KPOINTS、POSCAR、POTCAR、CHGCAR五个文件,其中CHGCAR文件是复制于静态自洽文件夹。
NOTE:根据不同的体系,可以自定义INCAR的参数,即自定义user_incar_settings={}这个字典。
接下来,我们讲解如何用Pymatgen进行态密度后处理,使用案例为Fm-3m的MgO结构。
计算文件夹包含:MgO_relax、MgO_static、MgO_dos
DFT计算得到的所有态密度信息都储存在vasprun.xml文件中,因此整个处理过程只需要解析vasprun.xml文件即可:
导入对应的包:
# 导入包
from pymatgen.io.vasp.outputs import Vasprun
import os
# 定义当前路径为工作路径
work_dir = '/work/home/andyhox/pymatgen_tutorials'
# 解析vasprun.xml文件
vasprun = Vasprun(os.path.join('MgO_dos','vasprun.xml'))提取TDOS数据:
# 提取dos数据
dos = vasprun.complete_dos
# 体系总态密度
tdos = dos.densities[Spin.up]对于没有自旋的体系,tdos的数据[Spin.up]即为所有的数据;如果是有磁性的体系,[Spin.up]和[Spin.down]分别表示自旋向上和自旋向下的态密度。在这里MgO没有磁性,在计算时设置ISPIN=1,因此提取的[Spin.down]即为所有数据。
from pymatgen.io.vasp.outputs import Vasprun
from pymatgen.core.periodic_table import Element
import pandas as pd
import os
work_dir = '/work/home/andyhox/pymatgen_tutorials'
# 提取dos数据
vasprun = Vasprun(os.path.join(work_dir,'MgO_dos','vasprun.xml'))
dos = vasprun.complete_dos
# 体系总态密度
tdos = dos.densities[Spin.up]
# 将dos的能量拟合到费米能级
energy = dos.energies - dos.efermi
# Mg元素总态密度
Mg_dos = dos.get_element_dos()[Element("Mg")].densities[Spin.up]
# O元素总态密度
O_dos = dos.get_element_dos()[Element("O")].densities[Spin.up]
dos_df = pd.DataFrame({
'Energy': energy,
'tdos': tdos,
'Mg': Mg_dos,
'O': O_dos
})
print(dos_df)打印输出为:
Energy tdos Mg O
0 -44.572532 0.0 0.0 0.0
1 -44.514432 0.0 0.0 0.0
2 -44.456232 0.0 0.0 0.0
3 -44.398032 0.0 0.0 0.0
4 -44.339832 0.0 0.0 0.0
... ... ... ... ...
1995 71.476068 0.0 0.0 0.0
1996 71.534268 0.0 0.0 0.0
1997 71.592368 0.0 0.0 0.0
1998 71.650568 0.0 0.0 0.0
1999 71.708768 0.0 0.0 0.0
[2000 rows x 4 columns]元素轨道态密度PDOS可以通过以下方法得到:
from pymatgen.io.vasp.outputs import Vasprun
from pymatgen.core.periodic_table import Element
from pymatgen.electronic_structure.core import OrbitalType, Spin
import pandas as pd
import os
work_dir = '/work/home/andyhox/pymatgen_tutorials'
# 提取dos数据
vasprun = Vasprun(os.path.join(work_dir,'MgO_dos','vasprun.xml'))
dos = vasprun.complete_dos
# 将dos的能量拟合到费米能级
energy = dos.energies - dos.efermi
# 体系总态密度
tdos = dos.densities[Spin.up]
# Mg元素轨道分态密度
Mg_dos = dos.get_element_spd_dos(el=Element("Mg"))
Mg_s = Mg_dos[OrbitalType.s].densities[Spin.up]
Mg_p = Mg_dos[OrbitalType.p].densities[Spin.up]
# O元素轨道分态密度
O_dos = dos.get_element_spd_dos(el=Element("O"))
O_s = O_dos[OrbitalType.s].densities[Spin.up]
O_p = O_dos[OrbitalType.p].densities[Spin.up]
# Create DataFrame directly from arrays
dos_df = pd.DataFrame({
'Energy': energy,
'tdos': tdos,
'Mg_s': Mg_s,
'Mg_p': Mg_p,
'O_s': O_s,
'O_p': O_p,
})
print(dos_df)打印输出:
Energy tdos Mg_s Mg_p O_s O_p
0 -44.572532 0.0 0.0 0.0 0.0 0.0
1 -44.514432 0.0 0.0 0.0 0.0 0.0
2 -44.456232 0.0 0.0 0.0 0.0 0.0
3 -44.398032 0.0 0.0 0.0 0.0 0.0
4 -44.339832 0.0 0.0 0.0 0.0 0.0
... ... ... ... ... ... ...
1995 71.476068 0.0 0.0 0.0 0.0 0.0
1996 71.534268 0.0 0.0 0.0 0.0 0.0
1997 71.592368 0.0 0.0 0.0 0.0 0.0
1998 71.650568 0.0 0.0 0.0 0.0 0.0
1999 71.708768 0.0 0.0 0.0 0.0 0.0
[2000 rows x 6 columns]上述提取的态密度数值均可以保存至csv文件供自定义绘图:
dos_df.to_csv(os.path.join(work_dir,'MgO_dos.csv'), index=False)此时在工作目录下会生成一个MgO_dos.csv文件,可以自由采用origin或者matplotlib进行绘图。
除了保存数据进行二次绘图之外,Pymatgen还支持直接读取态密度进行绘图。此时不需要提取具体态密度的数值,直接调用DosPlotter方法出图。
代码如下:
# 导入包
from pymatgen.electronic_structure.plotter import DosPlotter
from pymatgen.electronic_structure.core import OrbitalType, Spin
from pymatgen.io.vasp.outputs import Vasprun
import os
# 定义当前路径为为工作路径
work_dir = '/work/home/andyhox/pymatgen_tutorials'
# 提取dos数据
vasprun = Vasprun(os.path.join('MgO_dos','vasprun.xml'))
dos = vasprun.complete_dos
# 实例化
dosplot = DosPlotter()
# Mg元素轨道分态密度
Mg_s = dos.get_element_spd_dos(el="Mg")[OrbitalType.s]
Mg_p = dos.get_element_spd_dos(el="Mg")[OrbitalType.p]
# O元素轨道分态密度
O_s = dos.get_element_spd_dos(el="O")[OrbitalType.s]
O_p = dos.get_element_spd_dos(el="O")[OrbitalType.p]
# 添加需要绘图的态密度轨道
dosplot.add_dos('tdos',dos)
dosplot.add_dos('Mg',Mg_s)
dosplot.add_dos('Mg_p',Mg_p)
dosplot.add_dos('O_s',O_s)
dosplot.add_dos('O_p',O_p)
dosplot.get_plot(xlim=(-6,8), ylim=(0,4))运行结果:
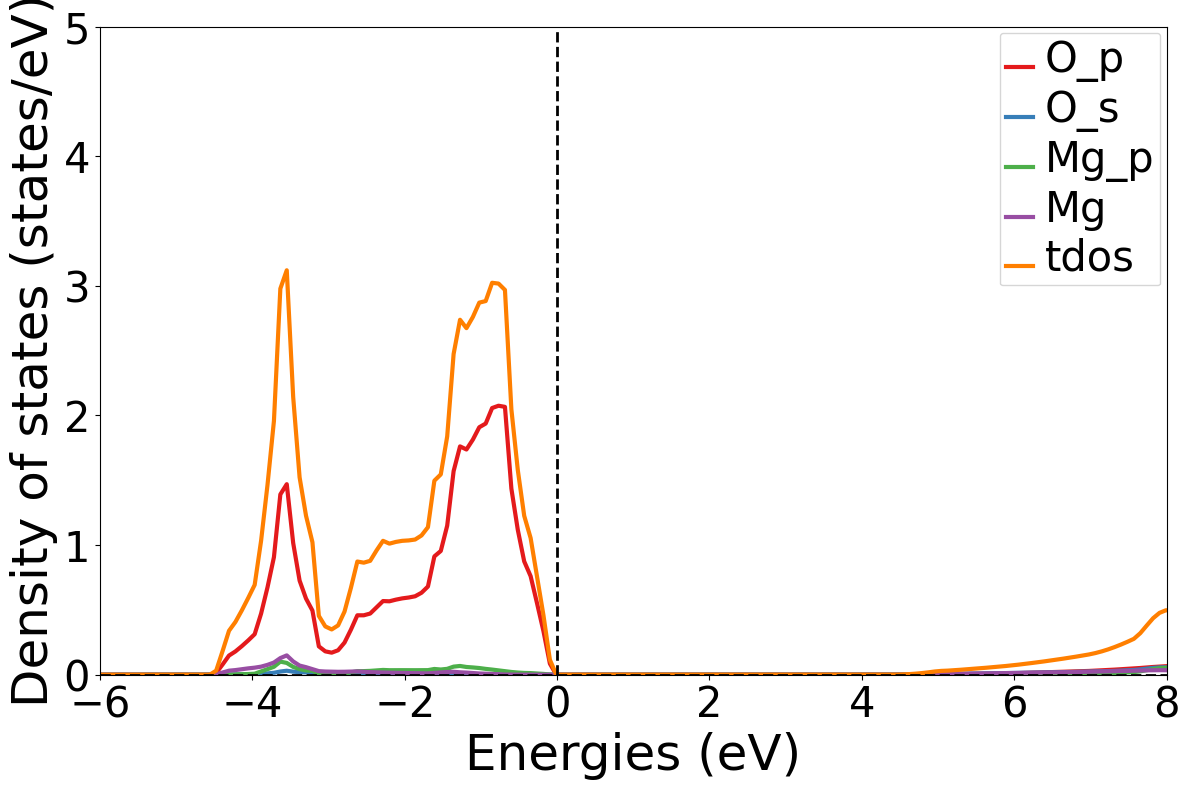
以上,我们就完成了在超算互联网使用Pymatgen实现材料电子性质计算生成输入文件、态密度处理及可视化的全流程,通过MPRelaxSet、MPStaticSet和MPNonSCFSet三个类方法显著提升了结构优化、静态自洽和非自洽计算的效率,为催化材料设计、半导体能带优化等场景提供了可复用的代码模板。