高性能计算服务
>
软件专区
>
最佳实践
>
GROMACS加速版:用两天时间快速估算油水体系的界面张力
在化学、材料和生物医药等领域,诸如乳液稳定性、三次采油及憎水材料等的界面张力研究中,分子动力学模拟已成为一种高效且精确的研究手段。而GROMACS作为一款高性能的开源 MD模拟软件,是相关科学研究和工程应用中的重要工具。
本期最佳实践,我们将对油-水体系的界面张力进行计算,此次分子模拟工作流设计思路是搭建油相模型——平衡——拉长模拟盒子——加入水分子——平衡——固定盒子形状模拟——计算界面张力。
基于异构加速卡,对约2万原子的体系进行1ns模拟,成本低至1元。
除了视频教程外,我们提供详细的实操文档,大家可参照这些步骤在超算互联网使用GROMACS进行多种体系界面张力的计算研究。
•界面张力的定义:增加不互相混合的临近两个相的界面尺寸所需的功,一般指液-液两相或者液-固两相,如果是气-液两相的界面,我们一般称为表面张力,而对于气-固界面,我们一般称为表面自由能。界面张力的单位是mN/m(毫牛每米),它表示每单位面积所需的功或每单位长度所需的力,用符号σ或者γ来表示。
•界面张力产生的原因:在相的边界区域,同一个相内部的分子之间的相互作用要比不同相中的分子的相互作用大,所以在界面位置的分子所受到的吸引作用就比相本体中的分子弱。在没有外力作用的情况下,物质就倾向于保持最小的相表面。如果想要增加界面的尺寸,则需要额外做功。
•研究界面张力的意义:许多领域都涉及界面张力的测量和研究,例如乳液稳定性研究、三次采油(Enhaced Oil Recovery, EOR)、憎水材料研究等。
•在进行模拟前需要明确所研究的问题:计算界面张力还是恒界面张力的模拟。如果是前者,根据文献中的描述方法如下:
表面张力(液体-真空界面)模拟可以采用纯液相盒子,将z方向长度扩大3倍,得到两个液体-真空界面,那么表面张力γ就可以从下式计算 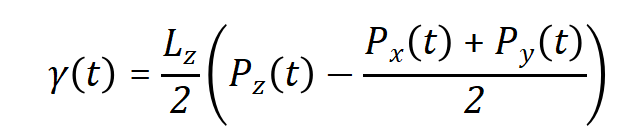 其中Pn是相应方向n的压强分量,而Lz是z方向(垂直于表面)的盒子长度。
其中Pn是相应方向n的压强分量,而Lz是z方向(垂直于表面)的盒子长度。
但是油-水界面不同于液体-真空界面,不能简单地把一个均相液体盒子的z方向维度拉长,而是需要添加不同种类的液体,并进行一段时间的平衡模拟。
如果是后者,进行恒界面张力的模拟,可以参考GROMACS手册的内容,在压强耦合算法一节当中,有pcoupltype=surface-tension的选项,其方法描述如下:
surface-tension压强耦合方式:当一个周期性体系包括至少两相,而且分隔面平行于xy平面,那么表面张力以及压强的z方向分量可以与恒压算法耦合。在GROMACS软件中,此功能需要使用Berendsen压强耦合算法。平均的表面张力γ(t)可以由法向方向和横向方向压强的差值进行计算,即 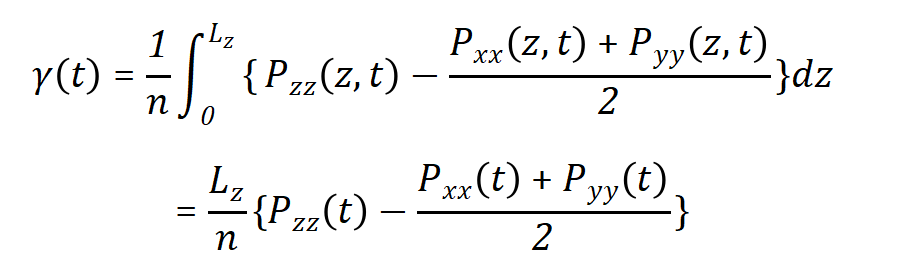 其中Lz是盒子的高度,n是界面的数量。z方向的压强使用Uzz调节盒子高度从而进行修正,即
其中Lz是盒子的高度,n是界面的数量。z方向的压强使用Uzz调节盒子高度从而进行修正,即 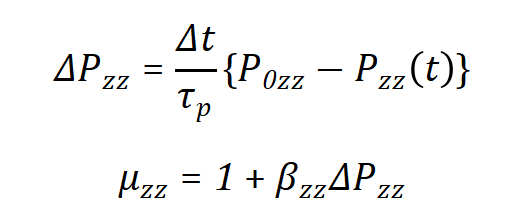
这与压强耦合算法相似,除了系数1/3是缺失的[1]。z方向的压强修正则用于使表面张力收敛,表面张力值相较于参考值γ0。在x/y方向上盒子的修正系数是 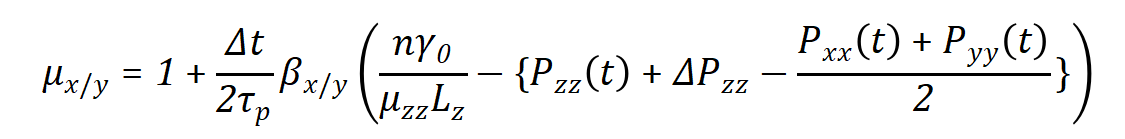
其中,βzz相比于其他压强耦合要求更严格,如果压缩率不准确,将会调节τp,然而表面张力耦合中它就会影响表面张力的收敛性。当βzz设定为零,即恒定盒子高度,那么ΔPzz也被设定为零,这对于得到准确的表面张力非常必要。
明确了本次油-水界面模拟属于前者,所以我们的分子模拟工作流设计思路应该是搭建油相模型——平衡——拉长模拟盒子——加入水分子——平衡——固定盒子形状模拟——计算界面张力。
•油-水层状模型选取了环己烷和水构成油水两相模型,其中环己烷液层位于模拟盒子中间,两侧加入水分子,预期整体模型如图1所示。 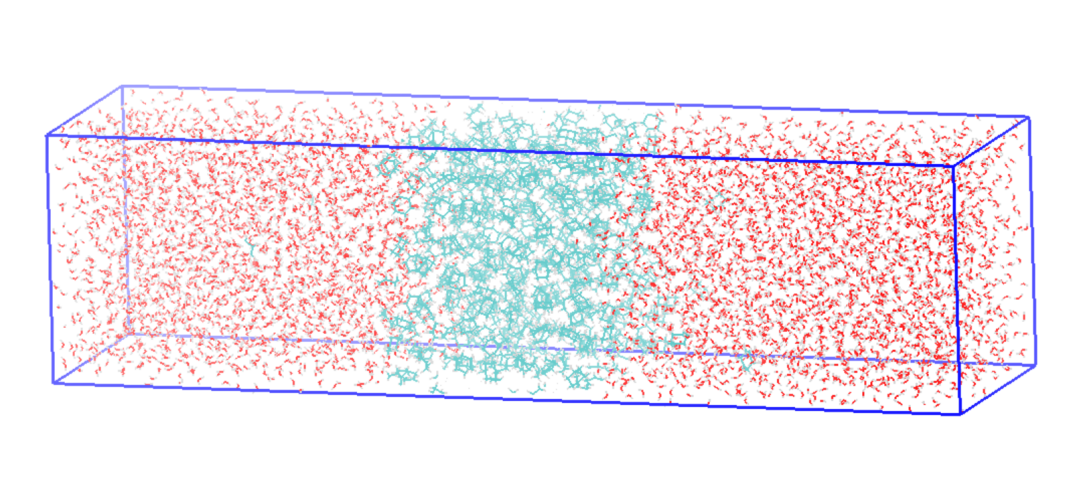
图1 油-水层状模型示意图,其中蓝色部分表示环己烷液层,红色部分表示水层。
•软硬件版本
本案例拟横向比较CPU并行版本和使用加速卡版本二者之间模拟速度的差别,因此选择了GROMACS v2024.1版本(CPU并行)和GROMACS v2023.2 加速版(异构加速卡)。 其中,CPU并行版将使用单节点7185处理器32核进行测试,异构加速版将使用7285处理器32核+4张加速卡(显存16GB HBM2)的异构节点进行测试。
第一天
我们可以选择例如LigParGen, ATB, AutoFF等工具构建环己烷分子的力场参数,这里我们选择OPLS-AA力场,假定我们已经准备了相应的*.itp和*.atp文件,同时我们需要准备一个环己烷分子的坐标文件C6R.gro用于构建油相液层,其中环己烷分子结构如图2所示。 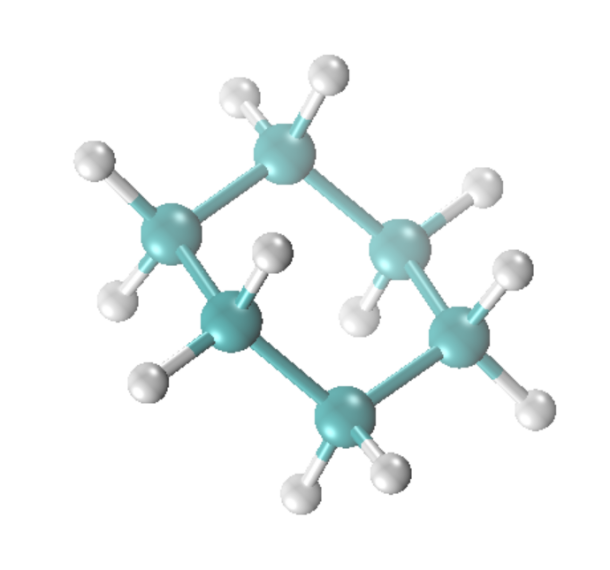 图2 环己烷分子的结构
图2 环己烷分子的结构
我们需要分步完成初始模型的搭建,其工作流如下: 在大盒子中插入环己烷分子——能量最小化——控温控压条件下压缩盒子以达到接近平衡的密度——控温控压模拟得到环己烷本体模型——拉长盒子z轴——在体系中加入水分子——能量最小化——获得油水液层模型。
按照上述工作流,我们先使用gmx_mpi insert-molecules -ci C6R.gro -box 4.8 4.8 4.8 -o oil_init.gro -nmol 400命令在边长4.8nm的立方体盒子中放入400个环己烷分子(为了确保可以放入400个环己烷分子,可以根据环己烷密度估算一下盒子的边长,再逐渐调节,不宜一次把盒子设置得过大),得到如下图所示。 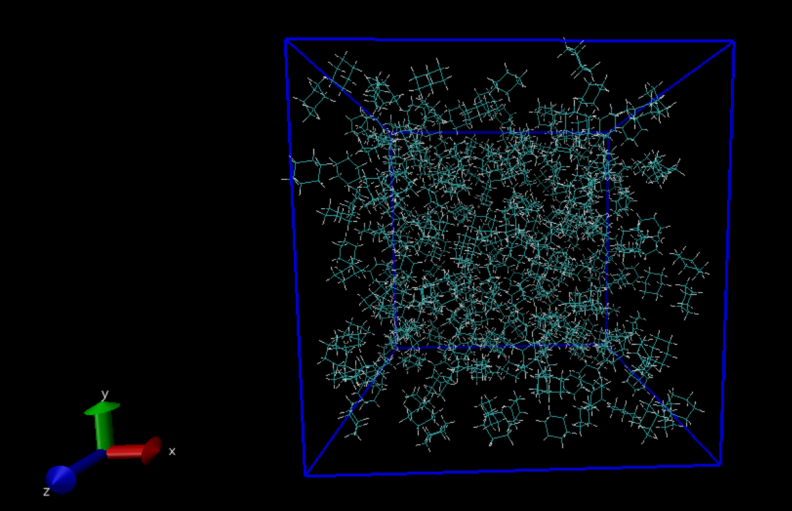 图3 使用insert-molecules插入环己烷分子的初始构象
图3 使用insert-molecules插入环己烷分子的初始构象
接下来将mdout.mdp文件里积分器设置为能量最小化integrator=steep,并修改提交任务脚本,能量最小化很快,并不需要加速卡资源,我们简单设置多cpu并行即可很快得到结果。得到的结构如下图所示。 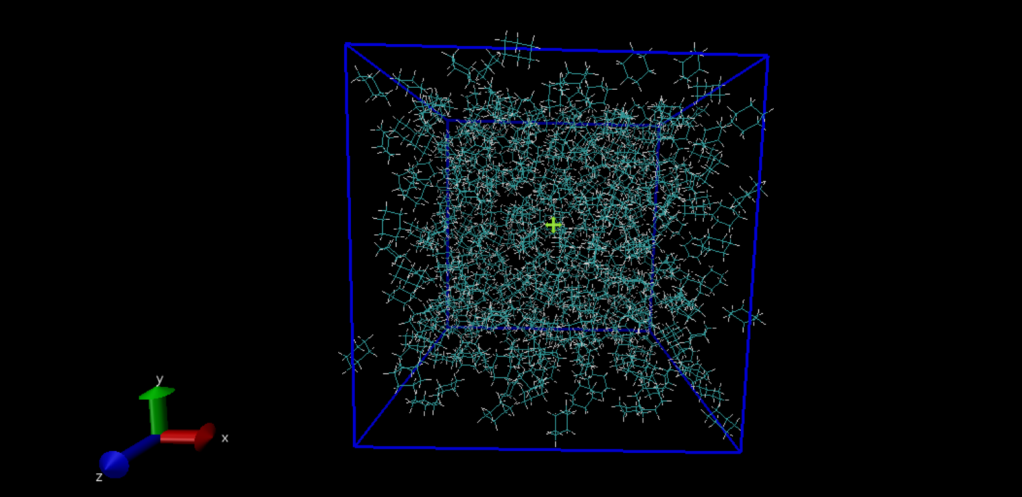 图4 能量最小化后的模拟盒子
图4 能量最小化后的模拟盒子
接下来进行控温控压模拟,需要修改mdout.mdp和提交任务脚本,其中mdp文件的修改关键是积分器修改成分子动力学,特别注意温度耦合和压强耦合,控温我们采用v-rescale方法,模拟时长可以根据盒子边长的变化情况来决定,压强耦合使用c-rescale方法。体系里包含7200个原子,数量不大,我们只需要使用8核并行即可,模拟完成得到环己烷本体模型,平均密度是764 kg/m3,如下图所示 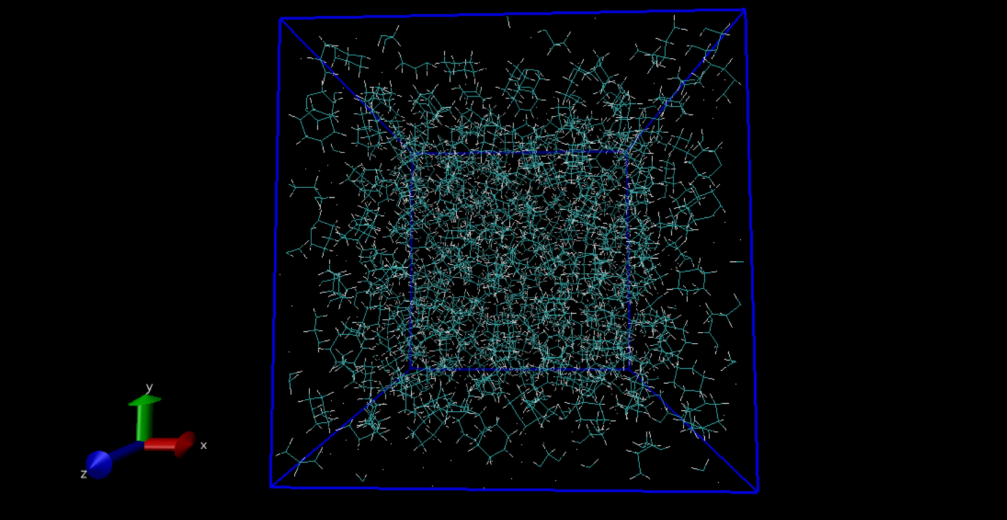 图5 环己烷本体模型
图5 环己烷本体模型
接下来我们使用gmx_mpi editconf -f bulk_oil.gro -box 4.18812 4.18812 12.56436 -o long.gro命令来拉长盒子,其中bulk_oil.gro是我们初步平衡后拿到的环己烷本体相的模型,在轴方向上延长了3倍,输出文件为long.gro,如下图所示。  图6 拉长盒子后的效果图
图6 拉长盒子后的效果图
接下来我们在盒子剩余的部分加入水分子,使用的命令是gmx_mpi solvate -cp long.gro -cs spc216.gro -box 4.18812 4.18812 12.56436 -radius 0.35 -o oil-water.gro,可以看到填充了水分子之后的效果,如下图所示。  使用这个功能可以免除分别做油层、水层的步骤,但是环己烷层中会混入许多水分子,这与我们的预期不符,所以我们可以使用自己熟悉的编程语言来简单处理一下,通过z坐标判据,去删掉混杂在环己烷层中的水分子,如下图。
使用这个功能可以免除分别做油层、水层的步骤,但是环己烷层中会混入许多水分子,这与我们的预期不符,所以我们可以使用自己熟悉的编程语言来简单处理一下,通过z坐标判据,去删掉混杂在环己烷层中的水分子,如下图。 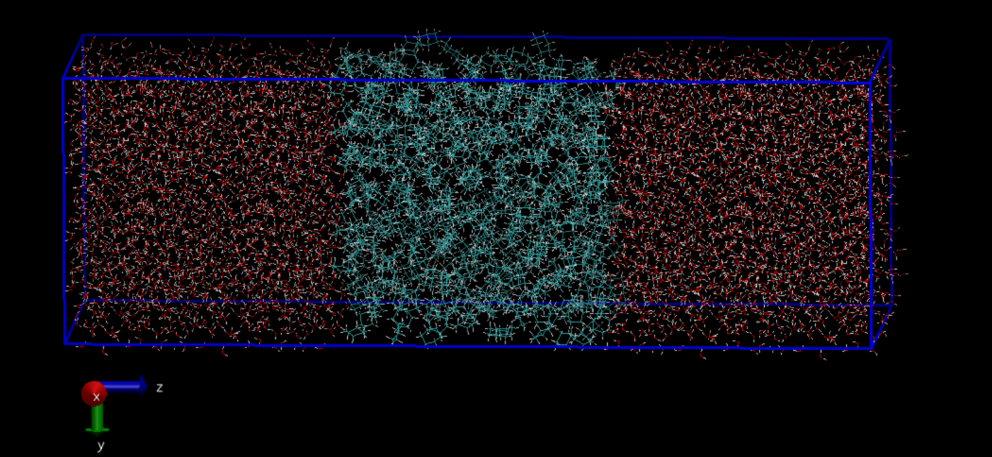 图7 去除多余水分子后的油-水层状模型
图7 去除多余水分子后的油-水层状模型
接下来进行能量最小化,需要修改力场参数文件,加入水分子的参数,即在topol.top文件中加入水分子的力场itp文件以及在分子个数加上SOL 水分子的数量。确保水分子数量和删除油层中水分子后的坐标一致。
现在模型中原子数比较多,我们可以考虑采用加速卡来提高模拟速度,需要注意的是,因为本文用的是MPI版本,不是线程MPI版,如果仅仅将非键作用放到加速卡计算,使用一张卡即可,如果是非键作用和静电相互作用都放到加速卡上计算,那么至少要用两张卡。预平衡阶段先用v-rescale控温和c-rescale控压让模型结构弛豫,然后再用v-rescale控温和半各向同性的parrinello-rahman控压方法模拟,达到平衡状态。下面是调用加速卡的脚本,需要注意的是参数的设置和其背后的原理。可以在长时间模拟前跑1ns测试一下加速效率和费用,数据对比详见末尾列表。
#!/bin/bash
#SBATCH -J gromacs-dcu
#SBATCH -p xahdnormal
#SBATCH -N 1
#SBATCH --ntasks-per-node=4
#SBATCH --cpus-per-task=8
#SBATCH --gres=dcu:4
module purge
source ~/apprepo/gromacs_MPI/2023.2-dtk23.10_hpcx241/scripts/env.shmpirun -np 4 gmx_mpi mdrun -pin on -s topol.tpr -ntomp 7 -nb gpu -gpu_id 0123 -npme 1 -pme gpu //4张卡把32个核分成了4组,每组需要一个mpi进程,每个组内有omp线程7个,非键相互作用放到gpu上去算,gpu的id号是0123,静电相互作用放到gpu上去算,其中有一组8cpu+加速卡是处理静电相互作用的 平衡模拟我们设置为10ns,预计需要2个半小时。平衡模拟结束后,修改mdp文件,固定盒子的体积,进行NVT模拟,可以设置模拟时长为10ns,得到轨迹文件后,就可以计算界面张力了。 让gmx_mpi mdrun跑起来,安心睡觉
第二天
GROMACS提供了丰富的数据分析工具,这里主要是使用gmx_mpi energy,其他的功能可以参考软件的使用说明,为了对比不同时间长度的界面张力的收敛性,我们可以比较一下不同统计时长(比如1ns, 2ns, 5ns, 8ns, 10ns)的界面张力的数据,使用gmx_mpi energy -b 开始的时间步 -e 指定的终了时间步就可以实现,界面会提示下列信息,如图所示 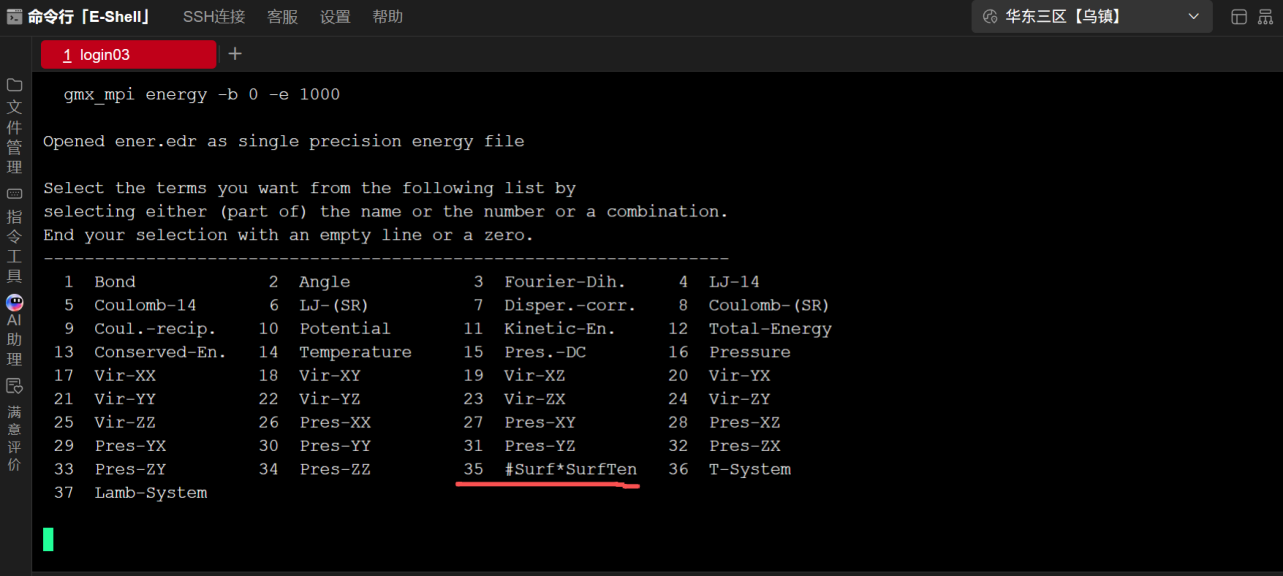 图8 选取界面张力统计数据
图8 选取界面张力统计数据
选择第35项,程序就会自动统计。由于统计的#Surf*SurfTen的单位是bar nm,换算成mN/m要除以10,而且体系当中有两个油水界面,所以需要除以20,那么就可以得到界面张力大约是23.5mN/m。将不同统计时间长度得到界面张力值对时间作图,如下图所示,数据趋势大体上可以得到界面张力的数值,至此,估算界面张力的任务就完成了。后台查看机时费用,两天不超过40元。 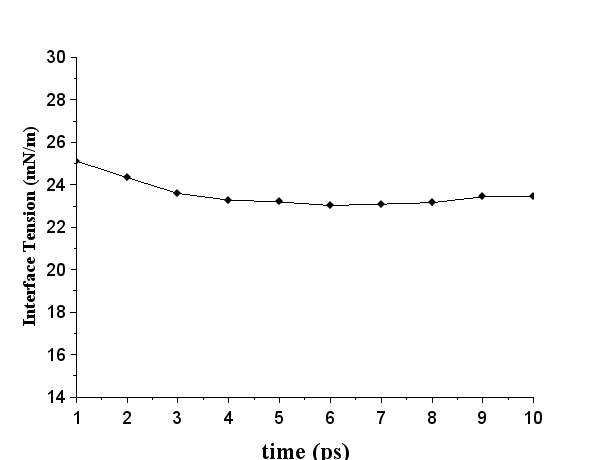 图9 界面张力-统计时间图
图9 界面张力-统计时间图
选取平衡后的液层模型,大约2万个原子,使用不同的算力配置跑1ns的NVT模拟,比较其计算速度和费用。 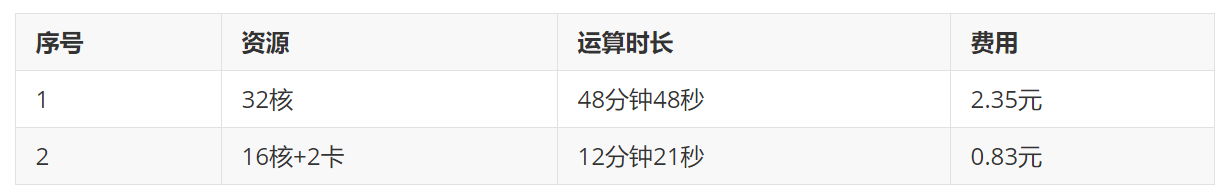
机时费对比: 预平衡模拟,体系中~2w的原子数,采用32核+4卡,16核+2卡,其完成1ns模拟所花费的时间和费用列表 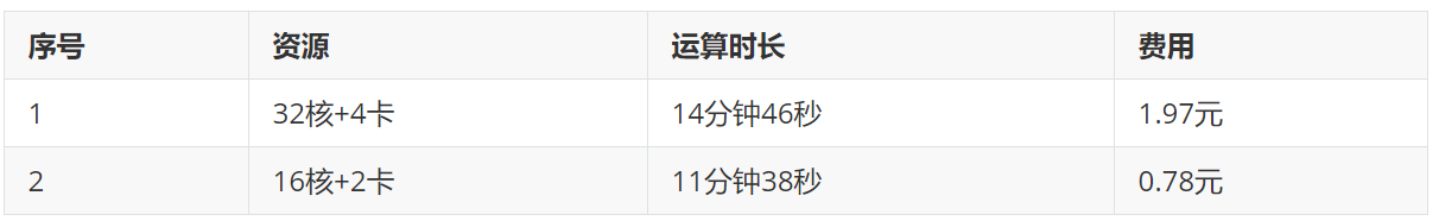 所以后续可以选择16核+2卡的算力配置。
所以后续可以选择16核+2卡的算力配置。
注意事项:
界面张力的结果受多方面因素影响,包括力场参数、模型大小、所选用的水分子模型等,本算例中为了快速估算,平衡时间和采样时间都相对比较短,建议在正式研究时进行基准验证和使用较长的平衡时间。
[1]根据GROMACS手册中压强耦合部分Berendsen标度法的说明,因为Berendsen标度方法可以各向同性的情况,所以就意味着不用把压强矩阵P的每个元素都给出来,而是可以使用一个对角矩阵,每个元素都是矩阵的迹除以3。对于有界面的体系,半各向同性标度是比较有用的。在这种情况下,在xy方向上是各向同性标度的,而z方向是独立标度的。如果想要在某一个方向上进行标度调节,可以把xy方向或者方向的压缩率设置为零。