高性能计算服务
>
软件专区
>
最佳实践
>
复现《Nature》19000℃黄金不熔化!LAMMPS进行激光加载的仿真与思考
近两万度高温,黄金都没熔化!这是发表在《Nature》期刊上的一项研究。超快激光在飞秒尺度内将金加热到远超理论极限,它“来不及”熔化,保持固态。这完全颠覆了40年前物理学家提出的“熵灾难”理论,“物理学不存在了?”
LAMMPS是一款用于分子动力学模拟的开源软件,广泛应用于材料科学、物理、化学和生物领域。它能够对各种粒子体系进行大规模的并行计算,适用于研究材料的原子级结构、分子行为等。
本次实操,我们将复现《Nature》正刊上发表的激光加载金薄膜的研究模拟,详细介绍如何在超算互联网使用ttm/mod模块进行激光加载模型,使用 LAMMPS 软件研究超高升温速率加载下金属体系的固液转变过程,以及是如何颠覆传统“熵灾难”假设的。 论文链接:https://www.nature.com/articles/s41586-025-09253-y
论文链接:https://www.nature.com/articles/s41586-025-09253-y
由于该论文中没给出激光加载中Au的各种参数,可以参考另一篇文献的研究结果,且经过试算验证可以正常计算。文献链接为https://doi.org/10.34133/2022/9754131
参数位于supplementary内。详细参数由下文代码部分给出。
其余仿真工况完全与论文一致,使用飞秒激光将金加载至15倍熔点,约19000℃后,金薄膜仍然维持固态的现象,颠覆了之前熵灾难假设,这里着重提取仿真细节,大约4.9J/cm2激光密度,加载45fs,3ps后仍保持固态,这就是我们需要的所有仿真参数。
建立室温单晶Au模型,使用ppp边界条件,X方向入射激光对整个模型进行加载,加载时间为0.045ps,加载结束后弛豫至10ps观察其结构与温度变化。
ttm/mod模块需要两个加载文件,分别是in文件与mod文件,in文件为通用的输入文件,mod文件为ttm/mod模块所必须的参数文件,in文件与mod文件完整代码如下,已做完整注释。
in 文件如下:
units metal # 使用 metal 单位制(Å, ps, eV, K)
atom_style atomic # 原子模型,不包含电荷或分子结构
boundary p p p # x、y、z 方向均采用周期性边界条件
shell "mkdir dump" # 创建用于存放 dump 文件的目录
variable latc equal 4.08 # 定义晶格常数变量 latc(Å)
lattice fcc ${latc} # 使用 FCC 晶格并指定晶格常数
variable xmax equal 50 # 模拟盒 x 方向最大边界
variable xmin equal 0 # 模拟盒 x 方向最小边界
variable ymax equal 40 # 模拟盒 y 方向最大边界
variable ymin equal -40 # 模拟盒 y 方向最小边界
variable zmax equal 30 # 模拟盒 z 方向最大边界
variable zmin equal -30 # 模拟盒 z 方向最小边界
region sim_box block ${xmin} ${xmax} ${ymin} ${ymax} ${zmin} ${zmax} # 定义整个模拟盒区域
create_box 2 sim_box # 在模拟盒中创建 2 种原子类型
region atom_box block ${xmin} ${xmax} ${ymin} ${ymax} ${zmin} ${zmax} # 定义填充原子的区域
create_atoms 1 region atom_box # 在 atom_box 区域内生成 1 号原子类型
region fushequ block INF INF INF INF INF INF # 定义辐照/TTM 作用区域(整个空间)
#region fushequ block INF INF -3 3 -3 3 # 备用的局部辐照区域定义(已注释)
group fushequ region fushequ # 将辐照区域内的原子定义为 fushequ 组
set group fushequ type 2 # 将 fushequ 组中的原子类型设为 2
mass 1 55.845 # 原子类型 1 的质量(amu)
mass 2 55.845 # 原子类型 2 的质量(amu)
velocity all create 300 123456 mom yes rot yes dist gaussian # 初始化原子速度,对应 300 K,高斯分布
pair_style eam/alloy # 使用 EAM 合金势函数
pair_coeff * * Au_Norman_2012.eam.alloy Au Au # 指定 Au 的 EAM 势文件和元素映射
neighbor 2.0 bin # 邻居列表截断距离
neigh_modify every 5 delay 0 check yes one 10000 # 邻居列表更新频率与检查策略
fix 1 all nve # 对所有原子采用 NVE 时间积分
fix twotemp fushequ ttm/mod 1354684 Au.ttm_mod 50 10 10 set 300.0 outfile 100 T_out.txt # 对 fushequ 组启用 TTM 模型并输出电子温度
compute pe all pe/atom # 计算每个原子的势能
compute ke all ke/atom # 计算每个原子的动能
variable KB equal 8.625e-5 # 玻尔兹曼常数(eV/K,metal 单位制)
variable TEMP atom c_ke/1.5/${KB} # 由原子动能计算瞬时原子温度
compute 1 all stress/atom NULL # 计算每个原子的应力张量
compute v all voronoi/atom # 计算每个原子的 Voronoi 体积
compute sumv all reduce sum c_v[1] # 计算系统总体积
variable AtomicVolume atom c_v[1] # 定义原子体积变量
variable Atomicstressx atom c_1[1]/c_v[1]/10000 # 计算 x 方向原子应力(单位换算)
variable Atomicstressy atom c_1[2]/c_v[1]/10000 # 计算 y 方向原子应力(单位换算)
variable Atomicstressz atom c_1[3]/c_v[1]/10000 # 计算 z 方向原子应力(单位换算)
timestep 0.0001 # 时间步长为 0.0001 ps(0.1 fs)
thermo 100 # 每 100 步输出一次热力学信息
dump 1 all custom 100 dump/result.*.xyz id type x y z c_pe c_ke v_TEMP v_Atomicstressx # 输出原子坐标、能量、温度和应力
thermo_style custom step temp etotal f_twotemp[1] f_twotemp[2] # 输出步数、晶格温度、总能和电子相关量
thermo_modify format float "%20.16g" # 设置热力学输出的浮点数格式
run 100000 # 运行 100000 步模拟mod文件如下:
a_0, energy/(temperature*electron) units # 电子比热容多项式展开的 0 阶系数
-5.3e-4 # a_0 的数值
a_1, energy/(temperature^2*electron) units # 电子比热容多项式的一阶温度系数
2.37e-6 # a_1 的数值
a_2, energy/(temperature^3*electron) units # 电子比热容多项式的二阶温度系数
1.3e-8 # a_2 的数值
a_3, energy/(temperature^4*electron) units # 电子比热容多项式的三阶温度系数
-1.22e-10 # a_3 的数值
a_4, energy/(temperature^5*electron) units # 电子比热容多项式的四阶温度系数
1.75e-12 # a_4 的数值
C_0, energy/(temperature*electron) units # 电子比热容常数项(低温修正)
5.3e-4 # C_0 的数值
A, 1/temperature units # 电子比热或耦合项中的温度相关系数
5.41e-3 # A 的数值
rho_e, electrons/volume units # 自由电子数密度
0.059 # 电子密度数值(electrons/Å^3)
D_e, length^2/time units # 电子热扩散系数
444899 # 电子扩散系数数值
gamma_p, mass/time units # 电子-声子确定性耦合系数
0.1524 # gamma_p 的数值
gamma_s, mass/time units # 电子-声子随机耦合系数
182.0 # gamma_s 的数值
v_0, length/time units # 电子有效漂移速度
27.3 # 漂移速度数值
I_0, energy/(time*length^2) units # 激光峰值能流密度(约 5 J/cm^2)
69333.33 # 激光强度数值
lsurface, electron grid units (positive integer) # 左侧表面在电子网格中的位置
0 # 左表面索引
rsurface, electron grid units (positive integer) # 右侧表面在电子网格中的位置
50 # 右表面索引
l_skin, length units # 表面电子皮肤层厚度
4 # 表面厚度数值
tau, time units # 激光加载或脉冲时间尺度
0.045 # 脉冲宽度数值
B, dimensionless # 激光或能量沉积的无量纲修正因子
0 # 未启用修正项
lambda, length units # 激光在材料中的吸收/衰减长度
400 # 吸收深度数值
n_ion, ions/volume units # 离子数密度
0.05 # 离子密度数值
surface_movement: 0 to disable tracking of surface motion, 1 to enable # 是否跟踪表面运动
0 # 关闭表面运动跟踪
T_e_min, temperature units # 电子温度下限
0 # 电子温度最小值目前,超算互联网已上线Lammps各个主要版本,支持开箱即用。
考虑到本次算例可以使用GPU加速,那么首选就是使用GPU加速版的Lammps(Lammps 2Aug2023加速版),在超算互联网购买后,通过我的商品,在应用软件中找到或搜索该版本商品,点击“命令行”,即可快速使用Lammps软件。
将写好的lammps输入文件上传到超算互联网平台并提交计算。
计算完成之后,从超算互联网平台下载轨迹文件。
在正确运行完成in文件之后,我们可以得到轨迹文件,其中包含着原子坐标与计算得到的原子温度。再次在应用软件中搜索Ovito,通过作业启动Ovito的图形化窗口,记得命令中调用至少一张GPU,即可开启图形化的Ovito界面。
将计算得到的轨迹文件拖入Ovito即可进行可视化操作。 上图是10ps内的原子轨迹图,确实可以看出直至10ps都保持了相当的固体结构,没有完全非晶液态化,并且直接计算最终rdf也可以发现,此时峰已经被平滑的很厉害了,处于一种即将迅速液化的不稳定固态,也是近似于熵灾难假设的。
上图是10ps内的原子轨迹图,确实可以看出直至10ps都保持了相当的固体结构,没有完全非晶液态化,并且直接计算最终rdf也可以发现,此时峰已经被平滑的很厉害了,处于一种即将迅速液化的不稳定固态,也是近似于熵灾难假设的。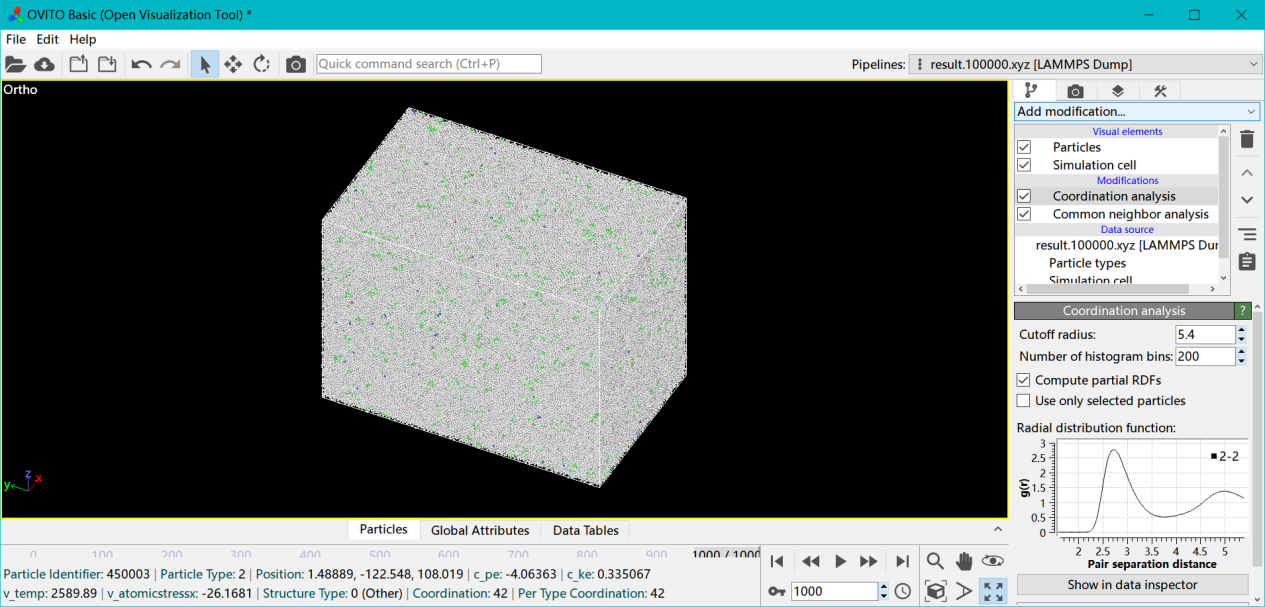 这里并没有提温度,因为在文献所给的激光强度和加载时间下,达不到那么高的晶格温度,甚至可以说相差甚远。
这里并没有提温度,因为在文献所给的激光强度和加载时间下,达不到那么高的晶格温度,甚至可以说相差甚远。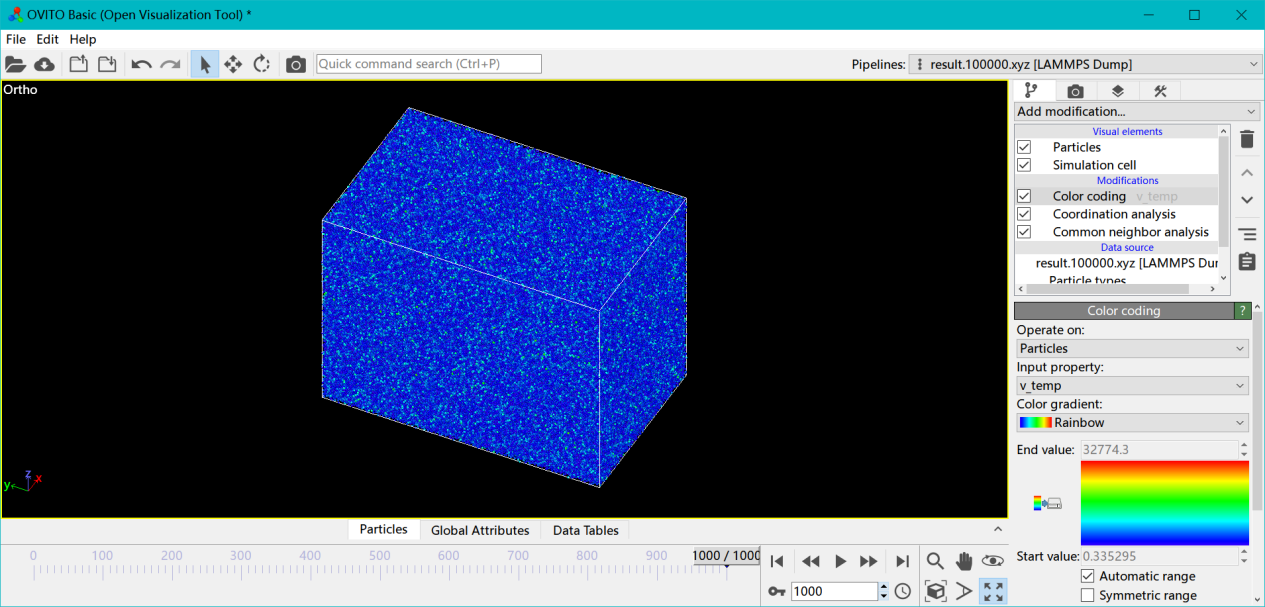 上图是未空间平均的图,下图是空间平均后的图,可以看出温度极值16000k,稍微平均一下就到熔点附近了,而激光加载本来就容易催生出局部超高温原子。
上图是未空间平均的图,下图是空间平均后的图,可以看出温度极值16000k,稍微平均一下就到熔点附近了,而激光加载本来就容易催生出局部超高温原子。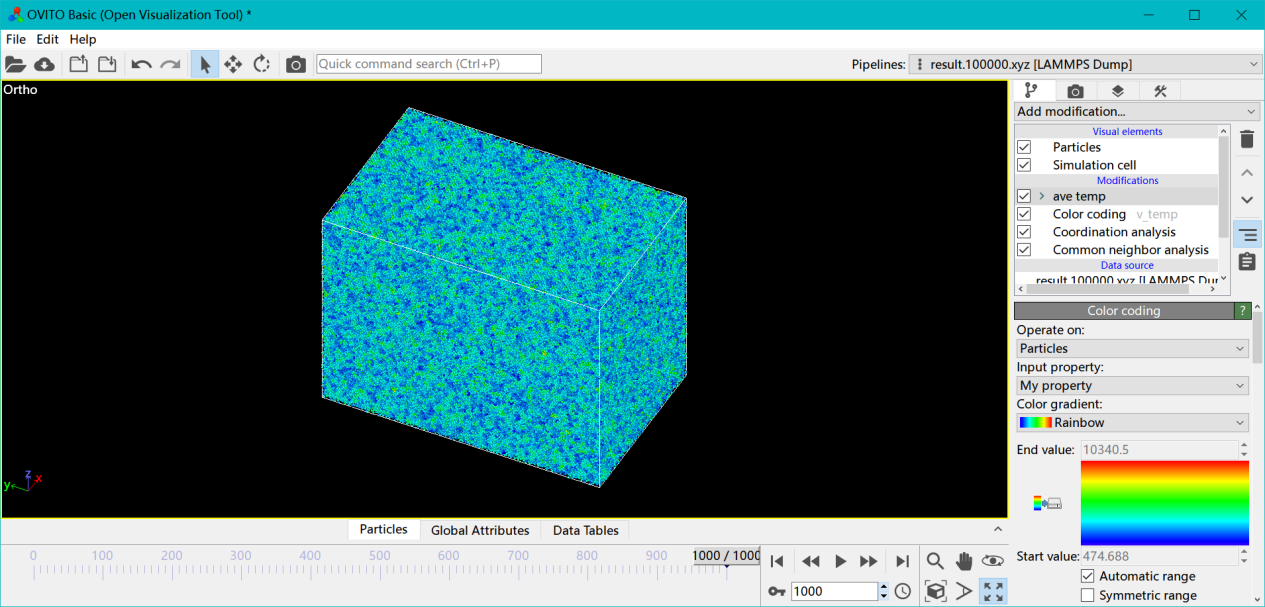 我们这里计算的是晶格温度,电子温度远超19000℃,这里文献里没太多涉及,就不进行研究了。
我们这里计算的是晶格温度,电子温度远超19000℃,这里文献里没太多涉及,就不进行研究了。
另一个比较微妙的点是,我在另一个激光密度更低、但加载时间更长的算例中,使用完全相同的势函数和材料参数,却得到了与之差距较大的结果。 这是加载区的剖面图,可以发现加载区显然已经无法保持固态了,局部高温区也更加明显,之后甚至发生了爆炸飞散,这里的加热速率就是如文献所说的1e15k/s,可结果却对不上。
这是加载区的剖面图,可以发现加载区显然已经无法保持固态了,局部高温区也更加明显,之后甚至发生了爆炸飞散,这里的加热速率就是如文献所说的1e15k/s,可结果却对不上。
唯一的区别就是这个算例加载区域较小,且总能量较大,且局部能量无法快速通过传热传递给周围晶格,形成了一个被其他晶格所约束的气化区,自然会产生极大的压力最终促使气化爆炸飞散,均匀加热中就不会出现这种状况。不过在这个算例中依然也算不出来19000℃的高温,试了试差不多35J/cm2才能做到,然而这时哪怕均匀加载也无法保持固态了。
本文并不是想否定这篇Nature文献的研究结果,实验部分的成果是很有价值的。
但值得注意的是,哪怕是nature,也完全可以通过相对合理的操作来让自己的仿真变得符合需求,我使用的Au参数可以通过更改吸收率或比热等,使其吸热能力大幅上升,使用的势函数也可以进行修正以适应超高温算例。真正有效可靠的,最终一定是实验,仿真的可靠性始终很差,在研究比较透彻的领域还勉强可信,在领域前沿就只能作为辅助工具了。
因此本期的最佳实践不仅仅是一次复现案例,更是希望能借这个案例提醒大家,仿真的可靠性与有效性极大的依赖于模型建立与计算细节,只有确保足够合理才能使自己的仿真更加可信。