高性能计算服务
>
>
GROMACS是一个开源且高效的分子动力学模拟软件,因其强大的功能和灵活性被广泛用于模拟蛋白质、核酸、脂质以及复杂生物体系的动态过程。GROMACS支持多种力场,允许用户自定义拓扑结构,并提供了丰富的分析工具来处理模拟数据。
本文,我们将介绍如何在超算互联网使用GROMACS软件,从原始的PDB结构文件开始,经过一系列系统构建、文件转化等步骤,最终执行mdrun进行生物分子模拟的一般过程。
使用GROMACS的mdrun命令进行分子动力学模拟,通常需要准备下述内容:
构建模拟系统: ①蛋白质/小分子/核酸的结构文件: 通常是 PDB 格式。
②添加溶剂: 使用 solvate 命令添加溶剂分子到模拟系统中。
③添加离子: 使用 genion 命令添加必要的离子以中和系统电荷。
分子动力学模拟文件: ①结构文件 (.gro): 包含分子系统的原子坐标。
②拓扑文件 (.top): 定义了分子的连接性、力场参数和其他相关细节。
③力场参数文件: 如 .itp 和 .ff 文件,这些文件定义了特定力场的参数。
④输入参数文件 (.mdp): 包含了模拟的具体参数设置,例如时间步长、模拟时间长度、温度耦合、压力耦合等。
PDB文件是一种广泛用于存储蛋白质、核酸、复杂组装体以及其他生物分子的三维结构数据的文件格式,在生物学、生物物理学、药物设计、结构生物学和教育等多个领域都有广泛应用。
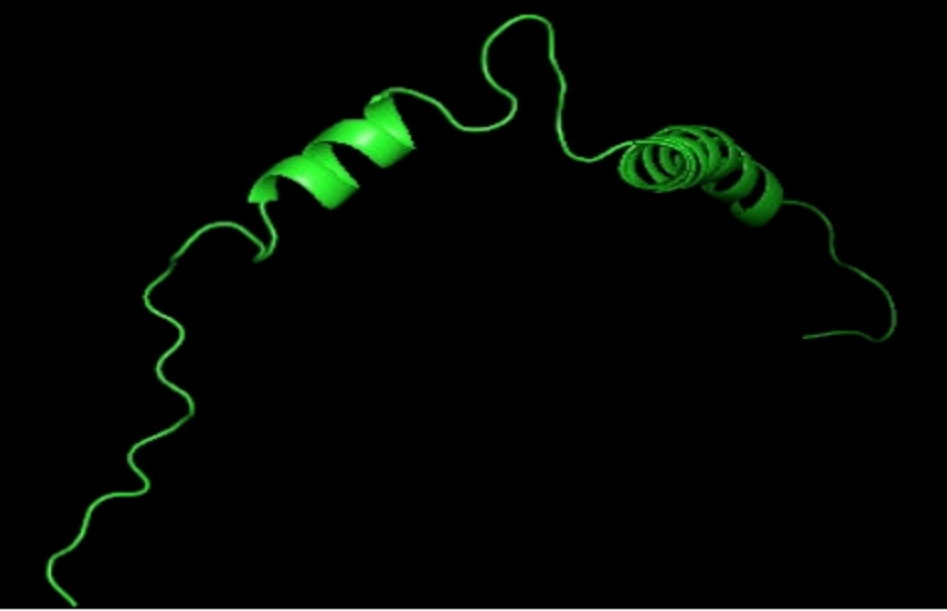
在GROMACS模拟中,PDB文件是模拟流程的起点,提供了初始坐标、分子结构等必要原始信息。实验使用的PDB测试算例来自蛋白质数据库RCSB。首先,我们从RCSB下载一个蛋白质结构,命名为input.pdb,其结构可使用如VMD、PyMOL等软件可视化展示如下:
超算互联网提供多版本、已编译的GROMACS软件,支持一键使用。本次实验使用gcc 9.3.0和intelmpi2017编译的GROMACS v2024.1版本。
您可以在超算互联网搜索“GROMACS v2024.1版本”购买软件,通过“我的商品”,在“应用软件”中找到“GROMACS v2024.1版本”,点击“命令行”,即可选择区域中心、命令行快速使用该软件。
准备好PDB文件和GROMACS软件后,接下来我们需要构建整个模拟系统。
在使用GROMACS软件进行分子动力学模拟时,从PDB文件生成 .gro 文件和 .top文件是准备模拟系统的一个重要步骤。gro文件定义了分子系统的几何结构,包括所有原子的位置,它通常被用来作为模拟的起始点。top文件包含了分子系统的化学拓扑信息,包括分子间的连接和相互作用等。
GROMACS提供了pbd2gmx实用程序,用于将蛋白质或其他大分子的 PDB 格式文件转换为.gro和.top文件。使用pdb2gmx时,程序会智能地搜索适合的力场数据库文件,这些文件包含了构建拓扑所需的详细信息,如原子类型、键合模式、非键相互作用参数等。您还可以通过命令行参数等方式指定力场,程序会从选定的力场目录中读取所有必要的文件。
实验使用的运行命令如下:
gmx_mpi pdb2gmx -f input.pdb -o input.gro -p topol.top -ff gromos54a7 -water spc –ignh其中,-f参数指定输入的pdb文件;-o参数指定输出的.gro文件名;-p参数指定输出的.top文件名;-ff参数指定使用的立场文件。GROMACS支持多种力场,如GROMOS、OPLS、AMBER等,您也可以使用自己的立场。本实验使用GROMACS软件包中的gromos54a7立场,它主要适用于生物大分子,尤其是蛋白质和核酸。–ignh参数忽略PDB文件中的所有氢原子,然后让pdb2gmx自动添加它们。
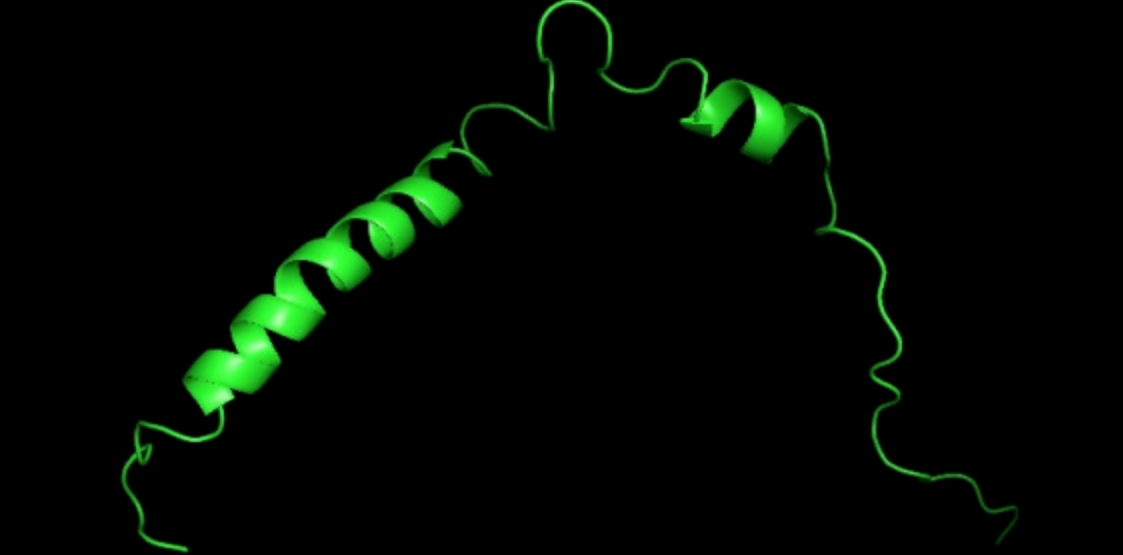
执行完毕pdb2gmx程序后,目录下会生成.gro文件和.top文件,以及对应的索引文件。我们可以使用可视化软件打开.gro文件,显示其结构。
创建模拟盒子是分子动力学模拟中的一个关键步骤,盒子定义了模拟中分子运动的空间范围,通常是一个具有周期性边界条件的三维盒子。盒子的大小和形状为分子的初始排布提供了框架,这对于模拟的准确性和可靠性至关重要。
GROMACS提供了editconf程序,用于处理和修改 .gro 文件,可通过选项 -box、-d 和 –angles创建和修改盒子的大小。
实验使用的运行命令如下:
gmx_mpi editconf -f input.gro -o input_box.gro -bt octahedron -d 1.0 -c在这个命令中,我们使用-bt参数指定生成八面体盒子,用于容纳模拟系统中的分子,-d参数指定溶质与盒子壁之间的距离为1纳米,新的结构文件命名为input_box.gro。
如果您想通过复制一个或多个分子来创建一个大的模拟系统,比如在研究脂质双层或非水溶剂时,可以使用GROMACS提供的“insert-molecules”程序来创建,本次实验暂不考虑。创建模拟盒子之后,整个系统的情况展示如下,可以看到蛋白质分子已经处于八面体中间位置。
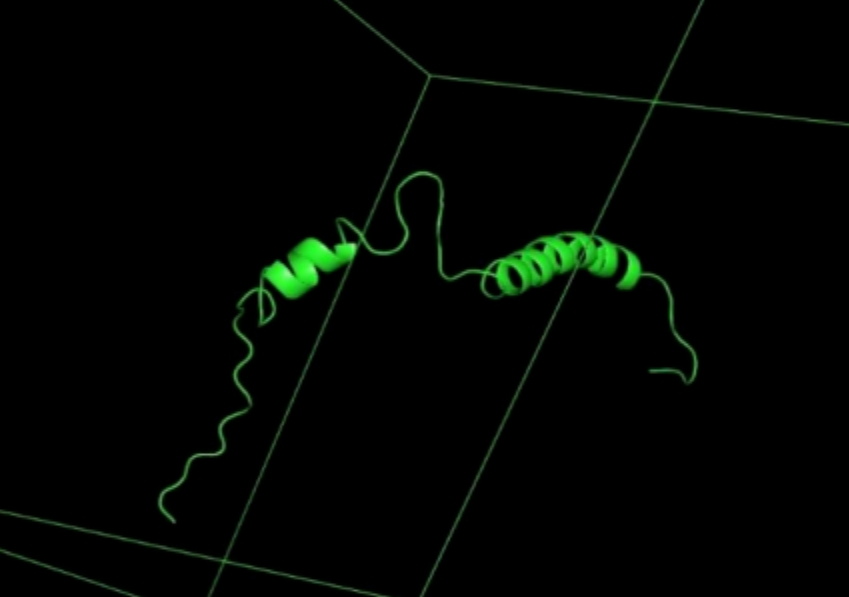
在分子动力学模拟中,添加溶剂确保了模拟环境能够准确地反映分子在实际条件下的行为。GROMACS 中的 solvate程序,能够在模拟盒子中添加溶剂分子,这个步骤会自动填充盒子,使其充满选定的溶剂模型。在添加溶剂之前,需要选择一个适合模拟体系的溶剂模型。GROMACS 支持多种溶剂模型,例如 SPC/E、TIP3P 等。
实验使用 solvate添加溶剂命令如下:
gmx_mpi solvate -cp input_box.gro -cs spc216.gro -p topol.top -o input_box_solvated.gro我们使用-cs参数指定了GROMACS自带的spc216.gro溶剂盒子来添加溶剂,并生成了新的input_box_solvated.gro文件。
添加了溶剂之后,整个系统的情况展示如下。
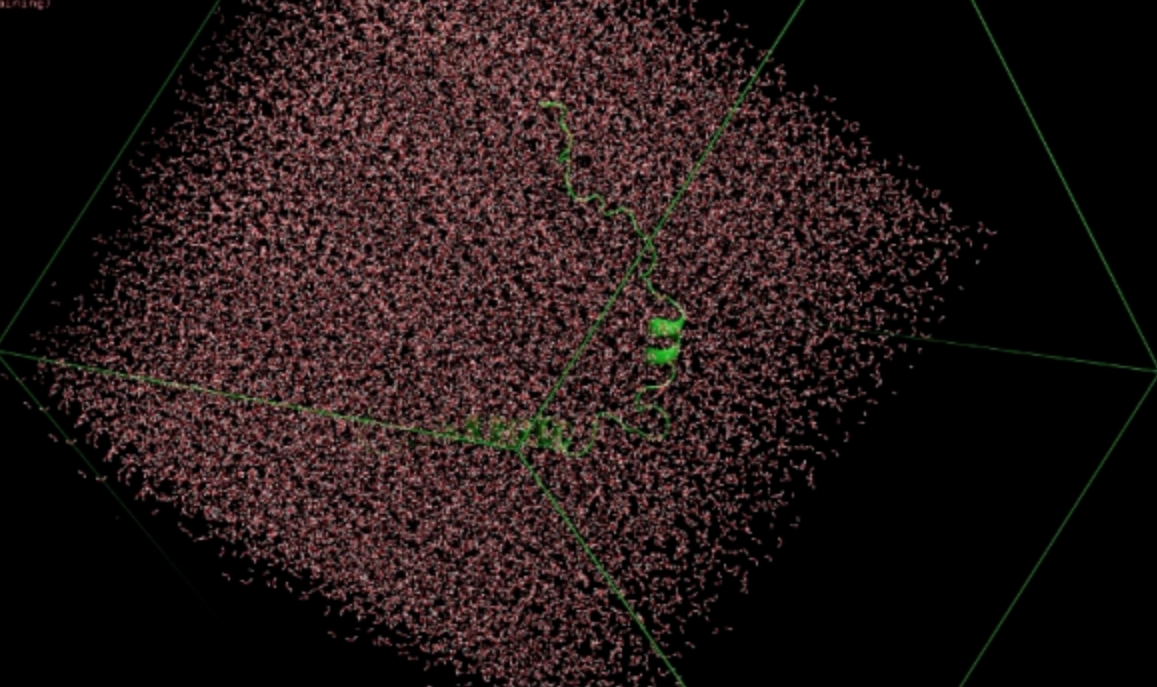
您还可以继续使用genion 工具,向系统中添加或移除离子,这个步骤可以中和系统的净电荷或者达到特定的离子浓度。检查添加离子后的系统是否电荷中性,确保模拟条件的准确性。在添加溶剂和离子后,进行能量最小化以优化系统的初始构型,进行一系列的平衡模拟,如 NVT 或 NPT 模拟,使系统达到热力学平衡。
通过这些步骤,您可以构建一个代表分子在溶液中行为的稳定模拟系统,为后续的分子动力学模拟做好准备。
.tpr 文件是 GROMACS 中的一种输入文件格式,称为运行输入文件(run input file)。它包含了分子动力学模拟所需的所有必要信息,包括原子坐标、速度、盒子尺寸、周期性边界条件、分子拓扑信息、力场参数以及模拟控制参数等。生成 .tpr 文件的一般步骤包括:
1、准备坐标和拓扑文件:这两个文件我们已经在上述步骤中生成。 2、创建或编辑 .mdp 文件:.mdp 文件包含了模拟的所有参数设置,如时间步长、温度控制、压力控制、模拟长度等。您需要根据模拟的需求创建或编辑这个文件。GROMACS也为我们提供了一些mdp文件,本文实验我们使用GROMACS测试算例中的system.mdp作为我们的mdp文件。 3、使用 gmx grompp 命令生成 .tpr 文件:
gmx_mpi grompp –f system.mdp -c input_box_solvated.gro -p topol.top -o input_box_solvated.tpr这里我们使用-f参数指定了包含模拟参数的.mdp文件,-c参数指定坐标文件,-p参数指定拓扑文件,以及-o参数指定了最终的输出文件。
生成的.tpr文件,可以使用gmx的check命令检查:
gmx_mpi check -c input_box_solvated.tpr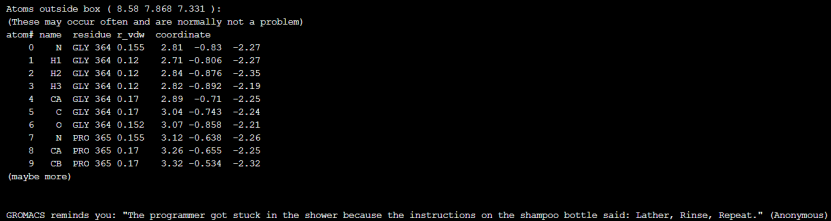
检查的输出信息告诉我们,有一些原子位于模拟盒子的边界之外,并列出了具体在盒子边界外的原子编号、名称、所属残基、范德华半径和坐标。GROMACS 提醒这种情况可能经常发生,并且通常不是问题,这是因为周期性边界条件允许原子在盒子的一边出去并在另一边重新出现。您在实际生成模拟系统的时候可以手动修正拓扑文件、坐标文件或模拟参数等,构建更加稳定的模拟系统。
mdrun是GROMACS中执行分子动力学模拟的核心工具。在配置好所有参数后,执行mdrun命令启动模拟。在模拟过程中,GROMACS会实时计算原子间的相互作用,并更新原子的位置和速度。
实验的运行命令如下:
gmx_mpi mdrun -ntomp 1 -v -s input_box_solvated.tpr -g test.log -e test.edr我们使用-s参数指定了最终生成的.tpr输入文件,通过 -v 参数,您可以获得更多的输出信息,这在调试或详细分析模拟时非常有用。提交作业运行结束后会输出负载分析,以及输出相关文件。
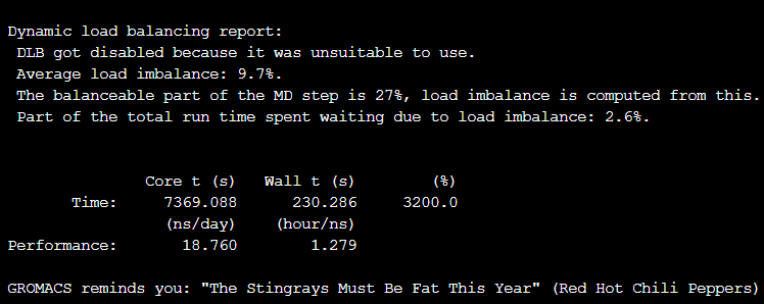
模拟产生的轨迹文件包含了原子在模拟过程中的位置和速度信息。通过分析这些数据,我们可以研究分子的动态行为和结构变化。除了轨迹分析,我们还可以从模拟中提取能量和动力学性质,如势能、动能、温度、压力等,以深入了解系统的热力学和动力学行为。
以上,我们就完成了从PDB文件到执行mdrun进行分子动力学模拟的一般过程。希望本篇最佳实践为您提供一些有价值的信息和实践技巧。